



ISSN 1757-8515
Utilising Therapeutic Hypothermia in the Control of Non-convulsive Status Epilepticus in a Patient with Creutzfeldt-Jakob Encephalopathy
Matthew J. Missert, Khalid J. Qazi and Catalina C. Ionita
Cite this article as: BJMP 2012;5(2):a515
|
|
Abstract Prion diseases are characterised by transmissible proteins which promote progressive neurological deterioration of their hosts. The most common prion disease is sporadic Creutzfeldt-Jakob disease (sCJD)1. Approximately 90 per cent of patients with CJD exhibit myoclonus at some point during their illness1. In a retrospective review of 1,384 patients with probable or definite CJD, 10 patients were admitted with an initial diagnosis of refractory non-convulsive status epilepticus (rNCSE)2. We encountered a patient with biopsy proven CJD who presented with rNCSE. Therapeutic hypothermia was instrumental in temporarily controlling the patient’s status epilepticus in combination with antiepileptic medications. Keywords: NCSE: Non-Convulsive Status Epilepticus; rNCSE: Refractory Non-Convulsive Status Epilepticus |
CASE PRESENTATION
A 67 year old Caucasian male presented to our institution with a one day history of uncontrollable movements. The patient was being evaluated by a psychiatrist, neurologist and a neuro-opthalmologist for a three month history of severe anxiety, gait instability and palinopsia, respectively. The patient had progressively worsened over the prior two weeks and at the time of presentation reported visual hallucinations with increased confusion. His involuntary movements escalated to the point where it appeared that he was having seizures.
His medical history was significant for gouty arthritis, hypertension, and major depression. His surgical history was notable for an open reduction and internal fixation of his left hip 6 years prior. There was no history of any blood or blood product transfusion. He was an insurance executive and did not have significant occupational exposures. His social and family history was unremarkable. His medications upon arrival included captopril, atenolol, bupropion, lamotrigine, clonazepam, folic acid, and ibuprofen.
On admission he was arousable, well nourished, afebrile, haemodynamically stable but disorientated. His cardiopulmonary, abdominal and integumentary examinations were unremarkable. Neurological examination was significant for bilateral symmetric hyperreflexia, diffuse cogwheel rigidity without a resting or postural tremor, and multi-focal dysrhythmic generalised myoclonus. No neck tenderness or nuchal rigidity was noted. A CT of the head without contrast in the causality department was negative for haemorrhage or any acute intracranial pathology.
His initial assessment showed confusion, hallucinations, myoclonus with medication induced delirium. Lewy body dementia, occipital lobe epilepsy, peduncular hallucinosis and prion disease were all considered in the differential diagnosis. On admission, laboratory data including a CBC, CMP, serum ammonia level, cardiac enzymes, urinalysis, and coagulation profile were unremarkable. A toxicology screen for illicit drugs and heavy metals was negative. The initial lumbar spinal fluid (CSF) analysis was only notable for mild proteinorachia of 57 mg/dL. Gram stain, India-ink stain, acid-fast bacilli (AFB), bacterial & fungal cultures, as well as PCR for viral nucleic acid (herpes, Varicella-Zoster, Epstein-Barr virus, arboviruses, and cytomegalovirus virus) were all negative. MRI with contrast was remarkable only for periventricular ischemic changes consistent with small vessel disease. The patient’s bupropion and lamotrigine were discontinued upon admission, and his clonazepam was increased with resolution of the myoclonus after 24 hours.
The admission EEG showed diffuse slowing with no epileptiform discharges or triphasic waves. Due to progressive neurologic deterioration, he was followed with serial electroencephalograms. On hospital day 5, he became unresponsive and the subsequent EEG revealed non-convulsive status epilepticus (NCSE). Temporary resolution of his seizures was achieved with lorazepam and pentobarbital infusions. After 3 days of almost complete suppression, the pentobarbital was discontinued without NCSE recurrence. On hospital day 15 the EEG again displayed NCSE. A ketamine drip was added to his drug regimen with only brief improvement. Pentobarbital had been restarted and progressively titrated up to the maximal dose without achieving burst suppression. Despite being on the maximal dose of pentobarbital, ketamine, valproic acid, levetiracetam, and topiramate he continued to display NCSE (Figure 1A).
At this point (hospital day 16), therapeutic hypothermia was initiated and continued for 48 hours. The patient’s core body temperature was maintained between 32-33 °C followed by slow rewarming to normothermia over the following 48 hours. Near complete suppression of epileptiform activity was observed on the EEG (Figure 1B). Ketamine and pentobarbital were successfully weaned off during the following days and phenobarbital was introduced without recurrence of NCSE.
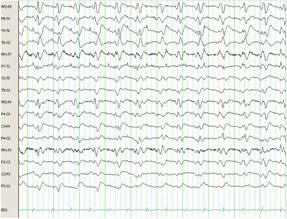
Figure 1A. Electroencephalogram from hospital day 15. Refractory non-convulsive status epilepticus while on ketamine, levetiracetam, valproic acid, topiramate and pentobarbital.
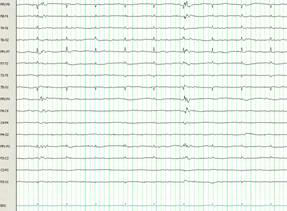
Figure 1B. Electroencephalogram from hospital day 16, after the initiation of therapeutic hypothermia. Suppression of epileptiform activity is observed after treatment with therapeutic hypothermia; ECG artifact persisting.
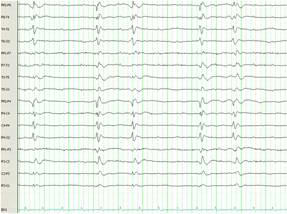
Figure 1C. Electroencephalogram from hospital day 29, 13 days after treatment with therapeutic hypothermia, illustrating generalised periodic sharp wave discharges with lack of background activity. Occasional triphasic waves are noted consistent with Creutzfeldt-Jakob encephalopathy.
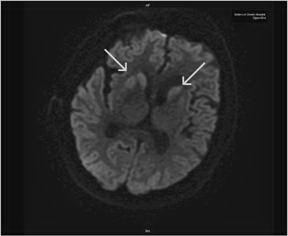
Figure 2. RepeatMRI of the brain on hospital day 21 illustrates asymmetric basal ganglia hyperintensities on diffusion weighted sequences, which are often observed in CJD.
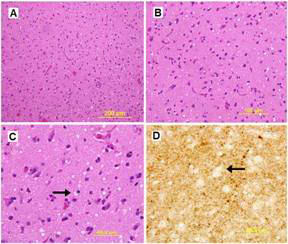
Figure 3. H & E stain of the cerebral cortex with low and high magnification (A & B). Coarse and fine vacuolization with spongiosis (arrows) are demonstrated on H & E and silver stain, respectively (C & D).
The patient had a repeat MRI which showed asymmetric basal ganglia hyper intensity on diffusion weighted imaging sequence consistent with CJD 3 (Figure 2)3. The results of CSF analysis for protein 14-3-3, neuron-specific enolase, and tau protein became available on hospital day 22. Despite controlling the NCSE, the patient remained unresponsive over the course of the following weeks. The EEG pattern changed to generalised periodic sharp waves, 1-2 per second with occasional triphasic waves, and a lack of background activity (Figure 1C). After fully reviewing the results with the family, an open brain biopsy was performed in effort to verify the diagnosis. The biopsy confirmed the diagnosis of spongiform encephalopathy (Figure 3). In light of the findings, withdrawal of care was initiated upon the family’s request and the patient expired on hospital day 42. The patient’s estimated symptomatic clinical course was approximately four and one-half months.
DISCUSSION
Creutzfeldt-Jakob disease is the archetype of prion mediated neurodegenerative disorders. There are 4 types of CJD; sporadic, familial, iatrogenic and variant4. The sporadic type accounts for 85 per cent of all cases of CJD4. The diagnosis of CJD and transmissible spongiform encephalopathy (TSE) can be elusive. The World Health Organisation’s diagnostic criteria for CJD require at least one of the following: (1) Neuropathological confirmation, (2) confirmation of protease-resistant prion protein (PrP) via immunohistochemistry or Western blot, or (3) presence of scrapie-associated fibrils4. However, newer and less invasive means for diagnosis have been explored in recent literature. CSF analysis for protein 14-3-3, tau protein, S100B protein and neuron-specific enolase have demonstrated sensitivities of 93 per cent, 89 per cent, 87 per cent and 78 per cent, respectively5. In addition, the use of MRI fluid-attenuated inversion recovery (FLAIR) and diffusion weighted imaging (DWI) techniques have yielded sensitivities of 91-92 per cent and specificities of 94 -95 per cent respectively, especially when utilised early in the disease state6. In our case, the initial MRI was unremarkable and only the repeated MRI, performed three weeks after admission, revealed basal ganglia signal intensities consistent with CJD.
One of the most studied and well characterised tools used to support the diagnosis of CJD is the EEG. The typical pattern observed in the early stages of CJD is frontal intermittent rhythmic delta activities (FIRDA). As the disease progresses, characteristic periodic sharp wave complexes (PSWC) can be observed, usually 8 to 12 weeks after the onset of symptoms7. However, the reported sensitivity of EEG is relatively low, ranging from 22 to 73 per cent, and is largely dependent of the subtype of CJD8. In our case, the patient presented with NCSE, which is an uncommon presentation of an uncommon disease. In a retrospective review of 1,384 patients with probable or definite CJD, only 0.007 per cent or 10 patients presented with NCSE2. Our patient did not demonstrate EEG findings consistent with CJD until late in his hospital course. Hence, CJD must be considered as a diagnosis in a patient who presents with refractory non-convulsive status epilepticus without overlooking the more common causes9.
The last important observation is the potential utility of therapeutic hypothermia in patients with refractory NCSE. Therapeutic hypothermia has long been known to suppress epileptiform discharge10,11. However, the safety and efficacy have not been broadly studied in human subjects. Corry and colleagues conducted a small study examining the effects of therapeutic hypothermia on 4 patients with refractory status epilepticus. The results were promising in that therapeutic hypothermia was successful in aborting seizure activity in all 4 patients and effectively suppressed seizure activity in 2 of the 4 patients after re-warming12. We were able to observe similar result in achieving temporary resolution of NCSE with therapeutic hypothermia in combination with antiepileptic medication in our patient.
|
Competing Interests None declared Author Details Matthew J. Missert, DO Sisters of Charity Hospital,Internal Medicine Training Program, Internal Medicine Resident PGY3,2157 Main Street, Buffalo, NY, 14214. Khalid J. Qazi, MD, Sisters of Charity Hospital, Professor of Medicine 2157 Main Street, Buffalo, NY, 14214. Catalina C. Ionita, MD, Sisters of Charity Hospital, Division of Neuroscience 2157 Main Street, Buffalo, NY, 14214. CORRESPONDENCE: Matthew J. Missert DO, Sisters of Charity Hospital, Internal Medicine Training Program Internal Medicine Resident PGY3, 2157 Main Street, Buffalo, NY, 14214. Email: matthew.missert@gmail.com |
References
1. Prusiner SB, Miller B. Prion Diseases. In: Harrison’s Principles of Internal Medicine, 17th edition, single edition. New York: McGraw-Hill. 2008:2646-2651.
2. Lapergue B, Demeret S, Denys V, et al. Sporadic Creutzfeldt-Jakob disease mimicking nonconvulsive status epilepticus. Neurology. 2010; 74:1995-1999.
3. Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004; 163:443-449.
4. World Health Organisation recommended standards and strategies for surveillance, prevention and control of communicable diseases: Creutzfeldt-Jakob disease (CJD) and variant CJD (vCJD). www.who.int/entity/zoonoses/diseases/Creutzfeldt.pdf
5. Collins SJ, Sanchez-Jaun P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278-2287.
6. Young GS, Geschwind MD, Fischbein NJ, et al. Diffusion weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. American Journal of Neuroradiology. 2005;26:1551-1562.
7. Wang PS, Wu YT, Hung CI, Kwan SY, Teng S, Soong BW. Early detection of periodic sharp wave complexes on EEG by independent component analysis in patients with Creutzfeldt-Jakob disease. Journal of Clinical Neurophysiology. 2008;25:25-31.
8. Brown, P. Transmissible spongiform encephalopathy in the 21st century: neuroscience for the clinical neurologist. Neurology. 2008;70:713-722.
9. Bleck TP. Less common etiologies of status epilepticus. Epilepsy Currents. 2010;2:31–33
10. Orlowski JP, Erenberg G, Lueders H, Cruse RP. Hypothermia and barbiturate coma for refractory status epilepticus. Critical Care Medicine. 1984;12:367-372.
11. Lundgren J, Smith ML, Blennow G, Siesj¨o BK. Hyperthermia aggravates and hypothermia ameliorates epileptic brain damage. Experimental Brain Research. 1994; 99:43-55.
12. Corry JJ, Dhar R, Murphy T, Diringer MN. Neurocritical Care. 2008; 9(2):189-197.

The above article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.