



ISSN 1757-8515
Integrative model of chronically activated immune-hormonal pathways important in the generation of fibromyalgia
Paul C. Breeding, Nancy C. Russell and Garth L. Nicolson
Cite this article as: BJMP 2012;5(3):a524
|
|
Abstract Clinicians are often challenged by patients presenting with a syndrome of chronic and diffuse full body pain with long standing fatigue and a cluster of related symptoms. Fibromyalgia has become the commonly accepted term for this syndrome. Diagnosis is established through recognized subjective symptoms, such as tender points and other indicators of chronic full body pain and fatigue. Suspected triggers have included bacterial and viral infections, toxins, allergens, and emotional and physical trauma. Unknown causes limit the prescription of effective treatments; however, neuropathic pain and fatigue have been identified as key components so dual reuptake inhibitors and anti-convulsants have shown some effectiveness for some patients. Based upon laboratory and clinical studies of the last decade, this article proposes a model for a subset of fibromyalgia patients who have prolonged immune activation with related oxidative and nitrogenous stress leading to multiple hormonal repression, disrupted collagen physiology, neuropathic pain and fatigue. This integrative model of fibromyalgia is based on chronic up-regulation of the immune system with subsequent hormonal, connective tissue and nervous system implications. |
Introduction
Fibromyalgia (FM) is a challenging set of chronic, overlapping and debilitating syndromes with widespread pain, abnormal pain processing, sleep disturbance, fatigue and psychological distress.1 The American College of Rheumatology (ACR) 1990 diagnostic guidelines were based primarily on tender point examination findings at 11 of 18 potential tender points;2 however, lack of consistent application of these guidelines in clinical settings led the ACR in 2010 to develop new diagnostic criteria based on a Widespread Pain Index (WPI) and symptom severity (SS) scale with no requirement of a tender point examination. Symptoms must have been present for at least three months with the absence of any other disorder that would otherwise explain the pain and other signs and symptoms.3
Type of pain and other symptoms vary widely in FM, complicating diagnosis and treatment. A cross-sectional survey of 3,035 patients in Germany utilized cluster analysis to evaluate daily records of symptoms noted by patients on handheld computers. Five subgroups were described: four with pain evoked by thermal stimuli, spontaneous burning pain, pressure pain, and pressure pain combined with spontaneous pain; the fifth subgroup had moderate sensory disturbances, but greater sleep disturbances and the highest depression scores.4
Estimates of the prevalence of FM have varied based on case definitions and survey methods. Using 1990 ACR guidelines, it was estimated to affect between 0.1 to 3.3% of populations in western countries and 2.0% in the United States. Greater prevalence occurs among females, with estimates ranging from 1.0 to 4.9%.1, 5 Reasons for the gender difference have not been determined.6-9
Fibromyalgia Risk Factors
Identification of risk factors for FM has been complicated by the array of seemingly unrelated signs and symptoms. The United States Centers for Disease Control (CDC) notes loose association with genetic predisposition,10 bacterial and viral infections, toxins, allergies, autoimmunity, obesity and both physical and emotional trauma.1, 11
Chronic fatigue syndrome and infection
Although chronic fatigue syndrome (CFS) has been defined as a separate syndrome, up to 70% of patients with FM are also diagnosed with CFS and 35-70% of patients with CFS have also been diagnosed with FM.12 Thus studies of patients with CFS may have clinical relevance to FM. Several case controlled studies of CFS and one of CFS/FM have been associated with chronic bacterial infections due to Chlamydia (Chlamydophila p.), Mycoplasma, Brucella, and Borrelia.12-18 The most prevalent chronic infection found has been that of the various Mycoplasmaspecies.15-23
Mycoplasmas are commonly found in the mucosa of the oral cavity, intestinal and urogenital tracts, but risk of systemic illness occurs with invasion into the blood vascular system and subsequent colonization of organs and other tissues.15-23 Mycoplasmal infections have been identified in 52 – 70% of CFS patients compared with 5 to 10% of healthy subjects in North America15-17, 19-22 and Europe (Belgium)23. For example, the odds ratio (OR) of finding Mycoplasma species in CFS was 13.8 (95% CL 5.8-32.9, p< 0.001) in North America.17 A review by Endresen12 concluded that mycoplasmal blood infection could be detected in about 50% of patients with CFS and/or FM. A CDC case-control study attempted to replicate these findings based on the hypothesis that intracellular bacteria would leave some evidence of cellular debris in cell-free plasma samples. Results were that the healthy subjects actually had evidence of more bacteria although the difference was not significant. The authors noted the complexity and limitations of this type of analysis and also postulated that since the CFS patients were years past the onset of illness, they might have previously cleared the triggering agent.24 However, most studies found Mycoplasma DNA in intracellular but not extracellular compartments in CFS patients, and this could explain the discrepancy.15-23 Other studies have found that 10.8% of CFS patients were positive for Brucella species (OR=8.2, 95% CL 1-66, p<0.01)16 and 8% werepositive for Chlamydia pn. (OR= 8.6; 95% CL 1-71.1,p< 0.01)17.
The presence of multiple co-infections may be an especially critical factor associated with either initiation or progression of CFS. Multiple infections have been found in about one-half of Mycoplasma-positive CFS patients (OR = 18.0, 95% CL 8.5-37.9, p< 0.001), compared with single infections in the few control subjects with any evidence of infection.17 A North American study identified chronic infections in 142 of 200 patients (71%) with 22% of all patients having multiple mycoplasmal infections while just 12 of the 100 control subjects (12%) had infections (p<0.01) and none had multiple infections.15 Similarly, a European study reported chronic mycoplasmal infections in 68.6% of CFS and 5.6% of controls. Multiple infections were found in 17.2% of the CFS patients compared with none in the controls (p<0.001).23 Multiple co-infections were also associated with significantly increased severity of symptoms (p<0.01).15, 23
Viral infections associated with CFS have included Epstein Barr virus, human herpes virus-6, cytomegalovirus, enteroviruses and several other viruses.15, 25, 26
Despite indications of single or multiple bacterial and/or viral infections in most patients with CFS, antibiotic or antiviral treatments have yielded inconsistent results.27 Slow growing intracellular bacteria are relatively insensitive to most antibiotics and have inactive phases when they would be completely insensitive to any antibiotics.28 23 Some treatments may actually have resolved the infections, but not the immune pathways that may remain in an activated state capable of producing symptoms.
Fibromyalgia and infection
Bacterial infections associated with FM as a separate syndrome have included small intestinal bacterial overgrowth (SIBO)29, 30 and helicobacter pylori (HP)31. Utilizing the lactulose hydrogen breath test (LHBT), investigators found SIBO in 100% of 42 patients with FM. They noted that 30-75% of patients with FM have also been found to have irritable bowel syndrome (IBS).29, 30 A confounding factor is that medications prescribed for FM often have gastrointestinal side effects.29 HP diagnosed by positive immunoglobulin gamma (IgG) serum antibody was significantly higher in women with FM (44/65 or 67.7%) compared with controls (18/41 or 43.9%) (p=0.025) in Turkey31.
Viral infections associated with FM have included hepatitis C, in which two studies found an association,32-34 and two studies found no association.35, 36 Associations with FM have also been found with hepatitis B, 37 human immunodeficiency virus (HIV)38, 39 and human T cell lymphotropic virus type I (HTLV-1).40
Fibromyalgia and non-infectious associations
Non-infectious triggers associated with FM have included toxins, allergens, and physical or emotional trauma. These triggers may not have been strictly “non-infectious” as allergens and toxins may also be produced by infections, and physical or emotional trauma may lead to the reactivation of previously controlled infections. Respondents to an internet survey of people with FM (n=2,596) also identified triggers as chronic stress (41.9%), emotional trauma (31.3%), acute illness (26.7%) and accidents (motor vehicle 16.1%, non-motor vehicle 17.1%).41 Physical trauma associated with FM has included cervical spine injuries as well as motor vehicle and other accidents.42-44
Fibromyalgia and autoimmunity
Three studies have found thyroid autoantibodies to be in greater percentages in subjects with FM compared with controls, in spite of normal thyroid hormone levels. One study reported autoantibodies in 41% of FM patients versus 15% of controls.45 The second study reported 16% in FM versus 7.3% in controls, p<0.01.46 The third study reported 34.4% in FM versus 18.8% in controls (p=0.025)47 and OR =3.87, 95% CL 1.54-10.13.48 This could also have been the result of thyroiditis, because infections like Mycoplasma are often found in thyroiditis patients.15
Autoantibodies to serotonin were identified in 74% of 50 patients with FM compared with 6% of 32 healthy (blood donor) controls. Notably, serotonin levels were normal in 90% of the FM patients indicating serotonin receptor involvement.49
Fibromyalgia and Metabolic Syndrome
Metabolic Syndrome consisting of abdominal obesity, high triglycerides, high blood pressure, elevated fasting glucose and decreased high-density lipids, was associated with FM in a U.S. study in which cases were 5.6 times as likely to have Metabolic Syndrome as controls (C2MH = 3.84, p = .047, 95% CL 1.25 – 24.74).50
Fibromyalgia and emotional trauma
Although emotional trauma has been acknowledged as a contributing factor, most studies of CFS/FM have used recognized tests such as Beck’s Depression Index, Beck’s Anxiety Index and Minnesota Multi Personality Index (MMPI) to exclude potential subjects with actual psychiatric illnesses.51, 52
Psychological and physiological subsets of fibromyalgia
A Wisconsin cross sectional survey of 107 women with confirmed diagnoses of FM used validated psychological and physiological measures followed by cluster analysis. Four distinct subsets were identified: (I) history of childhood maltreatment and hypocortisolism with the most pain and disability; (II) “physiological dysregulation” described as “distinctive on nearly every biological index measured” with high levels of pain, fatigue and disability; (III) normal biomarkers with intermediate pain severity and higher global functioning; and (IV) psychological well-being with less disability and pain.53
The “physiological dysregulation” of FM subset II consisted of the highest antinuclear antibody (ANA) titers (t=4.06, p=0.001), highest total cholesterol levels (t=3.96, p<0.001), larger body mass index (BMI) values t=2.21, p<0.04), lowest Natural Killer (NK) cell numbers (t=3.95, p<0.001), lowest growth hormone (t=3.20, p<0.002), and lowest testosterone levels (t=3.80, p<0.001). Trends were also indicated toward the highest erythrocyte sedimentation rate (ESR) (t=2.02, p=0.056), lowest creatinine clearance (t=1.85, p=0.067) and lowest cortisol (t=2.78, p<0.007).53
Proposed Model of Fibromyalgia
The authors’ proposed model of FM develops a rationale for the “physiological dysregulation” indicated in subset II of the Wisconsin study. In this model, various triggers are followed by prolonged immune activation with subsequent multiple hormonal repression, disrupted collagen physiology and neuropathic pain.
Activation of immune response pathways
Innate immune responses begin with anatomical barriers, such as the epithelium and mucosal layers of the gastrointestinal, urogenital and respiratory tracts, and physiological barriers, such as the low pH of stomach acid and hydrolytic enzymes in bodily secretions. 54 Breeching of these barriers activates cell-mediated immunity launched by leucocytes with pattern recognition receptors: neutrophils, macrophages and dendritic cells (DCs).54 Insufficient or damaged anatomical or physiological barriers would necessarily keep this cell mediated level of innate defense in a constant state of alert and activity.
In contrast to the innate immune response, adaptive immunity has highly specific recognition and response activities resulting in lasting changes produced by leukocytes known as lymphocytes. B lymphocytes (B cells) secrete plasma cells producing antibodies to specific pathogens. T lymphocytes (T cells), the other major cells of adaptive immunity, can be either cytotoxic (Tc) or helper cells (Th). Tc cells produce progeny that are toxic to non-self peptides and Th lymphocytes secrete small proteins (cytokines) that mediate signaling between leukocytes and other cell types. All types of lymphocytes retain memory so that subsequent invasions provoke faster and more rapid differentiation into effector cells. 54, 55 Some Th cells respond to intracellular pathogens (Th1) and some to extracellular pathogens (Th2). A third type (Th17) appears to respond to certain bacterial and fungal infections, tumor cells and are also involved in autoimmune diseases.56
In the presence of environmental stressors, cells may release stress proteins to alert the organism to potentially damaging conditions. These proteins can bind to peptides and other proteins to facilitate surveillance of both the intracellular and extracellular protein environment. One form of stress proteins, heat shock proteins (HSP), can mimic the effects of inflammation and can be microbicidal.52, 57
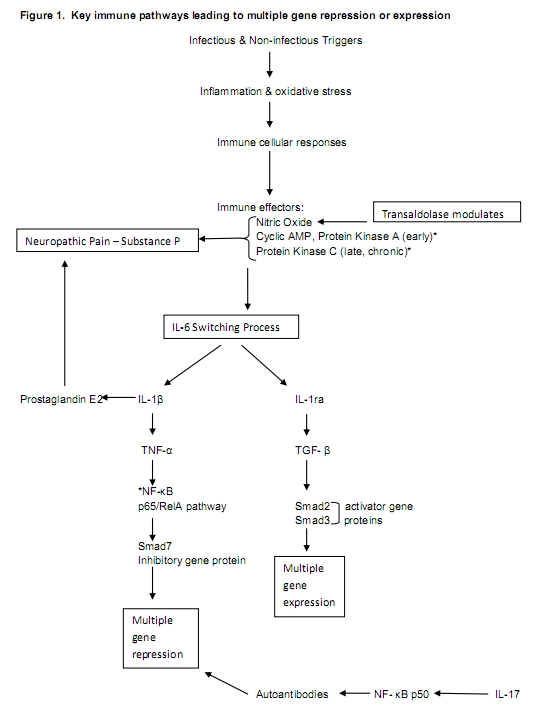
One of the earliest responses to intracellular viral or bacterial infections involves production of three types of interferon (IFNa, IFNb and IFNg). Any of these can initiate a series of metabolic events in uninfected host cells that produce an antiviral or anti-bacterial state.58, 59 When IFN-γ targets genes in uninfected cells, the targeted genes become microbicidal by encoding enzymes generating oxygen (O2) and nitric oxide (NO) radicals.58 Activation of O2 or NO radicals triggers another cascade involving IL-6, IL-1b, the cytokine Tumor Necrosis Factor-a (TNF-a) and the transcription nuclear factor kB (IKKb-NF-kB). NF-kB can be activated by a variety of inflammatory stimuli, such as cytokines, growth factors, hormones, oncogenes, viruses and their products, bacteria and fungi and their products, eukaryotic parasites, oxidative and chemical stresses, therapeutic and recreational drugs, additional chemical agents, natural products, and physical and psychological stresses.60 Activation of NF-kB releases its subunits; the p50 subunit has been associated with autoimmunity and the RelA/p65 unit with transcriptional activity involving cell adhesion molecules, cytokines, hematopoietic growth factors, acute phase proteins, transcription factors and viral genes.61 The authors propose that chronic infection or other stress would be a sustaining trigger of an immune cascade that includes NF-kB and resultant cell signaling processes that drive many of the symptoms of fibromyalgia.
The cytokine interleukin-6 (IL-6) can either activate or repress NF-kB through a switching mechanism involving IL-1ra and Interleukin 1b(IL-1b). IL-6 first activates Interleukin 1b (IL-1b), which then activates TNF-a, leading to the subsequent activation of NF-kB. 62, 63 Specifically, the release of the RelA/p65 subunit of activated NF-kB switches on an inhibitory signaling protein gene (Smad 7) that blocks phosphorylation of Transforming Growth Factor Beta (TGF-b) resulting in the repression of multiple genes. Alternatively, IL-6 activates IL-1ra, which allows TGF-b to phosphorylate and induce the expression of activating signaling protein genes Smad2 and Smad3, resulting in the full expression of multiple genes.61
NF-kB plays a key role in the development and maintenance of intra- (Th1) and inter- (Th2) cellular immunity through the regulation of developing B and T lymphocytes. The p50 dimer of NF-kB has been shown to block B Cell Receptor (BCR) editing in macrophages, resulting in loss of recognition and tolerance of host cells (autoimmunity). T cells that are strongly auto-reactive are normally eliminated in the thymus, but weakly reactive ones are allowed to survive to be subsequently regulated by regulatory T-cells and macrophages. Acquired defects in peripheral T-regulatory cells may mean failure to recognize and eliminate weakly reactive ones.54, 64 The IL-17 cytokine associated with autoimmunity can activate NF-kB through a pathway that does not require TNF-a.56 NF-kB activity can also be activated or repressed by the conversion of adenosine triphosphate (ATP) to cyclic adenosine monophosphate (c AMP) in the early phases (3 days) of nerve injury through its main effector enzyme, protein kinase A (PKA).65, 66 PKA decreases during later stages as the enzyme protein kinase C (PKC) increases. PKC then plays important roles in several cell type specific signal transduction cascades.67 An isoform of PKC within primary afferent nociceptive nerve fibers signals through IL-1b and prostaglandins E2 (PGE2) as demonstrated in animal studies.68 This process has been called “hyperalgesic priming,” and it has been described as responsible for the switch from acute to long-lasting hypersensitivity to inflammatory cytokines.69
Figure 1 depicts key immune pathways leading to expression or repression of multiple genes proposed to be important in FM and neuropathic pain.
Fibromyalgia and immune - hormonal interactions
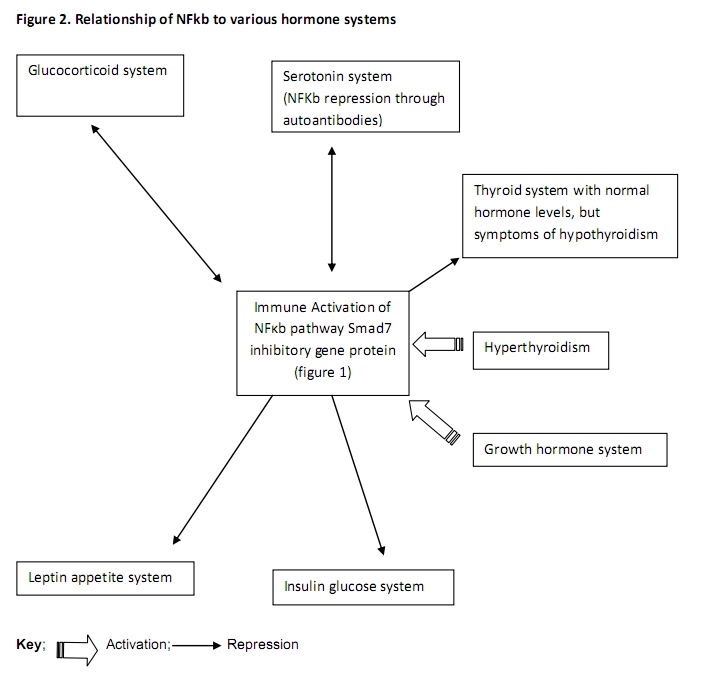
Reciprocity exists between the immune system and the hypothalamic-pituitary-adrenal (HPA) axis through its production of glucocorticoid signal transduction cascades. 63, 70, 71. Hormones such as cortisol (hydrocortisone) produced by the adrenal cortex, affect metabolism of glucose, fat and protein.72 The glucocorticoid receptor (GR), a member of the steroid/thyroid/retinoid super family of nuclear receptors is expressed in “virtually all cells”. When the GR in the cytoplasm binds a glucocorticoid, it migrates to the nucleus where it modulates gene transcription resulting in either expression or repression of TNF-a, IL-1bβ and the NF-kB p65/Rel A subunit. However, the RelA/p65 protein can also repress the Glucocorticoid Receptor. 63, 70, 71, 73
Growth hormone (GH), an activator of NF-kB,74 is usually secreted by the anterior pituitary, but changes found in FM may be hypothalamic in origin. GH is needed for normal childhood growth and adult recovery from physical stresses.75 Although low levels of GH were found in subset II of the Wisconsin study, 53 functional deficiency may be expressed as low insulin-like growth factor 1 (IGF-1) combined with elevated GH, suggesting GH resistance.76, 77 Defective GH response to exercise has been associated with increased pain and elevated levels of IL-1b, IL-6, and IL-8.77, 78
The hormones serotonin and norepinephrine modulate the movement of pain signals within the brain. Serotonin has been found to suppress inflammatory cytokine generation by human monocytes through inhibition of the NF-kB cytokine pathway in vitro;79 however, NF-kB promotion of antibodies can repress serotonin.49 Selective serotonin and norepinephrine reuptake inhibitors (SSNRIs), such as duloxetine and milnacipran, are key treatment options for fibromyalgia and have been approved of by the U.S. Food and Drug Administration (FDA).80, 81 Although serotonin has been best measured in cerebral spinal fluid (CSF), recently improved methods of collection were utilized (using rats and in 18 women) that yielded a high degree of correlation (r=0.97) between CSF and plasma, platelet, and urine measurements.82
NF-kB activation has also been documented to interfere with thyroid hormone action through impairment of Triiodothyronine (T3) gene expression in hepatic cells. 83 However, T3 administration has induced oxidative stress and activated NF-kB in rats.84
Metabolic Syndrome, a confounding factor in Fibromyalgia
Leptin and insulin hormones interact to regulate appetite and energy metabolism. Leptin, produced by adipose cells, circulates in the blood eventually crossing the blood-brain barrier to bond with a network of receptors within the hypothalamus. Insulin, produced by beta cells in the pancreas, similarly crosses the blood brain barrier to interact with its own network of hypothalamic receptors. Leptin and its receptors share structural and functional similarities to long-chain helical cytokines, such as IL-6, and it has been suggested that leptin be classified as a cytokine.85-89
Metabolic syndrome can be a confounding factor in FM due to peripheral accumulation of fatty acids, acylglycerols and lipid intermediates in liver, bone, skeletal muscle and endothelial cells. This promotes oxidative endoplasmic reticulum (ER) stress and the activation of inflammatory pathways involving PKC and hypothalamic NF-kB, leading to central insulin and leptin repression.85-87, 89-91 Hyperinsulinemia further stimulates adipose cells to secrete and attract cytokines such as TNFa and IL-6 that trigger NF-kB in a positive feedback loop, which can be complicated by chronic over nutrition that increases the generation of reactive oxygen intermediates and monocyte chemoattractant protein-1 (MCP-1).87, 89 When exposed to a chronic high fat diet, hypothalamic NF-kB was activated two fold in normal mice and six times in mice with the obese (OB) gene.89
Fibromyalgia and indicators of immune-hormonal activity
Although most components of either innate or adaptive cell mediated immune responses exist for only fractions of seconds, some of their effects and products can be detected long after in the skin, muscle, blood, saliva or sweat92, 93.
One component, nitric oxide (NO), can suppress bacteria; however, endothelial damage causes dysfunction with impaired release of NO and loss of its protective properties.86 The enzyme transaldolase acts as a counterbalance by limiting NO damage to normal cells. Thus, high levels of transaldolase indicate elevated reactive oxygen species, reactive nitrogen species (ROS/RNS) and cellular stress. The “exclusive and significant over-expression of transaldolase” in the saliva samples of 22 women with FM compared with 26 healthy controls (77.3% sensitivity and 84.6% specificity, p<0.0001; 3 times greater than controls; p=0.02) was “the most relevant observation”; although there was no correlation between transaldolase expression and the severity of FM symptoms.92
High levels of NO have been associated with high levels of insulin, and insulin itself is a vasodilator that, in turn, can stimulate NO production. Beta cells of the pancreas are quite susceptible to ROS/NOS damage .86 When free radical damage of beta cells reaches critical mass, insulin production plummets with an associated decline in NO levels. Thus, patients with FM who have high NO levels would likely be suffering from associated metabolic syndrome, and patients with low NO levels would likely be suffering from Type II diabetes.85, 88
Figure 2 illustrates the relationship of NF-kB to various hormone systems.
Fibromyalgia and immune-hormonal influences on connective tissue
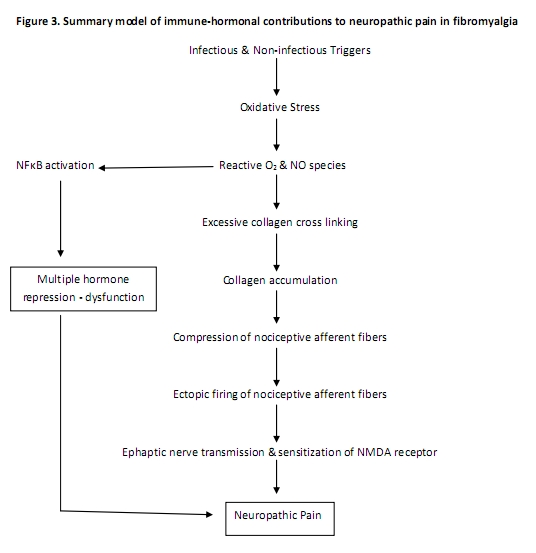
Inflammation of muscles, tendons, and/or fascia is generally followed by proliferative and remodeling phases of healing initiated by fibroblasts which lay down an extracellular matrix (ECM) composed of collagen and elastin fibers. “Fibroblasts respond to mechanical strain by altering shape and alignment, undergoing hyperplasia and secreting inflammatory cytokines including IL-6.” The extra cellular matrix is initially laid down in a disorganized pattern that is subsequently matured and aligned. Chronic and excessive mechanical tension from postural imbalance, hormonal disruption or other factors may interfere with collagen maturation. 94 Remodeling of the extracellular matrix and collagen deposition around terminal nerve fibers may be compressive and contribute to neuropathic pain.95
Oxidative stress in muscles accelerates the generation of advanced glucose (glycation) end products (AGEs). AGE-mediated cross-linked proteins have decreased solubility and are highly resistant to proteolytic digestion. Interaction of AGEs with their receptors leads to activation of NF-kB resulting in an increased expression of cytokines, chemokines, growth factors, and adhesion molecules.96 97
Two AGE products have been reported at significantly elevated levels in the serum of patients with FM: N-carboxymethyllysine (CML) (2386.56 ± 73.48 pmol/mL; CL 61.36-2611.76 versus controls 2121.97 ± 459.41 pmol/mL; CL 2020.39-2223.560; p<0.05)96 and pentosidine (mean 190 ± 120 SD and median 164 versus controls mean 128 ± 37 SD and median 124; p<0.05)97 Comparison of muscle biopsies showed “clear differences in the intensity and distribution of the immunohistochemical staining”. CML was seen primarily in the interstitial tissue between the muscle fibers where collagens were localized and in the endothelium of small vessels of patients. Activated NF-kB was seen in cells of the interstitial tissue especially around the vessels of patients, but almost no activated NF-kB was seen in the control biopsies. AGE activation of NF-kB has been shown to be significantly more prolonged than the activation of NF-kB by cytokines.96 97
Fibromyalgia, the nervous system and pain
Sensory transmission in humans occurs through three primary afferentnerve fiber types: heavily myelinated mechanical afferent pathways (A Beta fibers) that transmit non-noxious tactile sensations, small-diameter myelinated fibers (A Delta fibers) that transmit sharp pain, and small diameter unmyelinated fibers (C fibers) that transmit dull aching pain. The heavily myelinated non-pain Aβ fiber type has been shown to sprout axons that terminate on pain lamina in the posterior horn of the spinal cord resulting in the conversion of mechanical stimuli to pain. Within the brain, sensitization of the N-methyl D-aspartate (NMDA) receptors can amplify pain signals between the thalamus and the sensory cortex.67, 98
Chronic damage or excitation of nociceptive afferent fibers from compressive collagen deposition may develop into spontaneous (ectopic) firing oscillating at frequencies sufficient to initiate cross (ephaptic) excitation of sympathetic and sensory fibers (myelinated A-delta and non-myelinated C fibers) within the dorsal root ganglia (DRG) of the central nervous system.98 Normally, the DRG has little sympathetic innervation, but trauma can trigger sympathetic sprouting that forms basket-like structures within the DRG. Neurotrophins, in particular nerve growth factor (NGF), play an important role in sympathetic fiber sprouting of sensory ganglia in murine models. DRG can be reservoirs for latent viral infections such as Herpes Zoster, HIV and enteroviruses. In addition, the Borrelia species has been identified in a non-human primate model of Lyme disease. NGF also facilitates expression of Substance P (SP), a peptide neurotransmitter involved in the induction of the IL-6 - NF-kB pathway 60, 99, 100 and in the transmission of neuropathic pain.101, 102 SP has been found to be elevated in the cerebrospinal fluid of patients with FM in comparison to normal values,103 and control subjects.104
Summary and Conclusions
Chronic unresolved infection, trauma, and/or emotional stresses that trigger immune pathways with subsequent chronic hormonal and nervous system responses is proposed to perpetuate chronic neuropathic pain. Figure 3 provides a summary model of immune-hormonal contributions to neuropathic pain in fibromyalgia.
The ACR criteria and severity scales have defined fibromyalgia and The Wisconsin study has identified psychological and physiological subsets that are critical steps in its characterization. This type of testing could be further strengthened through the use of specific biomarkers. Potential markers of FM status include the RelA/p65 and p50 subunits of NF-kB, which are currently the focus of several clinical trials of other chronic painful conditions. Additional potential markers include: IL-6, IL-1b, TNF-a, PKC, transaldolase, CML, pentosidine and NGF. Substance P has been previously identified as a marker of pain, but is problematic as a marker for FM, since it has only been measured in the CSF. The search for markers that are truly specific to FM may continue to be a difficult task due to their overlap with other metabolic conditions, such as CFS, metabolic syndrome, type II diabetes, and IBS. Nonetheless, these markers remain important as they can indicate oxidative stress, cytokine activation, hormonal dysregulation and neuropathic pain. These potential FM markers need to be evaluated in clinical trials where they can be measured over time and correlated with patient symptoms.
Currently, family and general medical practice physicians are uniquely positioned to establish the FM diagnosis, determine subsets of FM patients, investigate potential triggers of chronic immune activation, advise patients, prescribe medications and refer patients to appropriate specialists or pain centers. Establishment of the FM diagnosis requires use of the ACR Widespread Pain Index (WPI) and symptom severity (SS) scale, but no longer requires the tender point examination. 3
Determination of FM subsets can be accomplished using the approach used in the Wisconsin cross sectional survey.53 Investigation of potential triggers of chronic immune activation needs to include sources of underlying infection, unresolved physical or emotional trauma, toxins and food sensitivities. These investigations may be accomplished through careful interviewing and well-designed questionnaires. Advising the patient should acknowledge the reality of their pain and other symptoms and provide rational approaches to resolution of those symptoms. Prescribing of medications needs to be sensitive to current and previous patient experience with medications, in addition to following current guidelines for stabilizing FM symptoms. Referral to appropriate specialists and centers would include those with expertise in physical medicine, psychology and nutrition. Physical medicine can address pain and functional deficits; psychology can address underlying emotional issues and trauma; and nutrition can focus on resolution of chronic inflammation, oxidative stress, and intestinal dysbiosis.
Where do we go from here for additional FM treatment options? Immune modulators have been used successfully in other painful conditions, such as rheumatoid arthritis. Immune modulators acting on the IL-6 - NF-kB cascade have considerable potential for FM, but only after ruling out or successfully treating any underlying infections. Numerous pharmaceutical blockers of NF-kB exist, but most are associated with serious side effects. Natural products may provide additional options as some are able to mediate pathways leading to NF-kB without the same side effects.105 Medications that elevate individual hormone levels have been included in accepted treatment protocols in the case of serotonin and norepinephrine. However, elevations of other hormones, such as cortisol and thyroid hormones, are under investigation and remain controversial. Elevation of individual hormones may be problematic because of the number of different hormones influenced by the IL-6 - NF-kB pathway.
|
Acknowledgements Paul Breeding proposed the initial model and wrote early drafts of the paper. Nancy Russell assisted in subsequent literature reviews and the writing of subsequent versions of the manuscript. Garth Nicolson contributed most of the section on infectious triggers and both critiqued and added to remaining parts of the manuscript. Competing Interests None declared Author Details GARTH L. NICOLSON, PhD President, Institute for Molecular Medicine, Department of Molecular Pathology, The Institute for Molecular Medicine, California, USA. PAUL C. BREEDING, DC Rehabilitation Specialist, The Institute for Molecular Medicine, California, USA. NANCY C. RUSSELL, DrPH Consultant, The Institute for Molecular Medicine, California, USA. CORRESPONDENCE: GARTH L. NICOLSON, PhD President, Institute for Molecular Medicine, Department of Molecular Pathology, The Institute for Molecular Medicine, P. O. Box 9355, S. Laguna Beach, CA 92652, USA. website: www.immed.org Email: gnicolson@immed.org |
References
1. Anonymous. Centers for Disease Control: Fibromyalgia 2012: Available from: http://www.cdc.gov/arthritis/basics/fibromyalgia.htm.
2. Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33(2):160-72.
3. Wolfe F, Clauw DJ, Fitzcharles MA, et al. Fibromyalgia criteria and severity scales for clinical and epidemiological studies: a modification of the ACR Preliminary Diagnostic Criteria for Fibromyalgia. J Rheumatol. 2011;38(6):1113-22.
4. Rehm SE, Koroschetz J, Gockel U, et al. A cross-sectional survey of 3035 patients with fibromyalgia: subgroups of patients with typical comorbidities and sensory symptom profiles. Rheumatology. 2010;49(6):1146-52.
5. Gran JT. The epidemiology of chronic generalized musculoskeletal pain. Best Pract Res Clin Rheumatol. 2003;17(4):547-61.
6. Yunus MB. Gender differences in fibromyalgia and other related syndromes. J Gend Specif Med. 2002;5(2):42-7.
7. Macfarlane TV, Blinkhorn A, Worthington HV, et al. Sex hormonal factors and chronic widespread pain: a population study among women. Rheumatology (Oxford). 2002;41(4):454-7.
8. Samborski W, Sobieska M, Pieta P, et al. Normal profile of sex hormones in women with primary fibromyalgia. Ann Acad Med Stetin. 2005;51(2):23-6.
9. Okifuji A, Turk DC. Sex hormones and pain in regularly menstruating women with fibromyalgia syndrome. J Pain. 2006;7(11):851-9.
10. Arnold LM, Hudson JI, Hess EV, et al. Family study of fibromyalgia. Arthritis and rheumatism. 2004;50(3):944-52.
11. Neumann L, Buskila D. Epidemiology of fibromyalgia. Curr Pain Headache Rep. 2003;7(5):362-8.
12. Endresen GK. Mycoplasma blood infection in chronic fatigue and fibromyalgia syndromes. Rheumatol Int. 2003;23(5):211-5.
13. Stratton CW, Sriram S. Association of Chlamydia pneumoniae with central nervous system disease. Microbes Infect. 2003;5(13):1249-53.
14. Chia JK, Chia LY. Chronic Chlamydia pneumoniae infection: a treatable cause of chronic fatigue syndrome. Clin Infect Dis. 1999;29(2):452-3.
15. Nicolson GL, Gan R, Haier J. Multiple co-infections (Mycoplasma, Chlamydia, human herpes virus-6) in blood of chronic fatigue syndrome patients: association with signs and symptoms. APMIS. 2003;111(5):557-66.
16. Nicolson GL, Gan R, Haier J. Evidence for Brucella spp. and Mycoplasma spp. co-infections in blood of chronic fatigue syndrome patients. J of Chronic Fatigue Syndrome. 2005;12(2):5-17.
17. Nicolson GL, Nasralla MY, De Meirleir K, et al. Evidence for Bacterial (Mycoplasma, Chlamydia) and Viral (HHV-6) Co-Infections in Chronic Fatigue Syndrome Patients. Journal of Chronic Fatigue Syndrome. 2003;11 (2):7-19.
18. Nicolson GL, Nicolson NL, Haier J. Chronic fatigue syndrome patients subsequently diagnosed with Lyme disease Borrelia burgdorferi: Evidence for Mycoplasma species co-infections. J of Chronic Fatigue Syndrome. 2008;14(4):5-17.
19. Nicolson GL, Nicolson NL. Gulf War illnesses: complex medical, scientific and political paradox. Med Confl Surviv. 1998;14(2):156-65.
20. Vojdani A, Choppa PC, Tagle C, et al. Detection of Mycoplasma genus and Mycoplasma fermentans by PCR in patients with Chronic Fatigue Syndrome. FEMS Immunol Med Microbiol. 1998;22(4):355-65.
21. Nasralla M, Haier J, Nicolson GL. Multiple mycoplasmal infections detected in blood of patients with chronic fatigue syndrome and/or fibromyalgia syndrome. Eur J Clin Microbiol Infect Dis. 1999;18(12):859-65.
22. Nasralla MY, Haier J, Nicolson NL, et al. Examination of mycoplasmas in blood of 565 chronic illness patients by polymerase chain reaction. Int J Med Biol Environ. 2000;28(1):15-23.
23. Nijs J, Nicolson GL, De Becker P, et al. High prevalence of Mycoplasma infections among European chronic fatigue syndrome patients. Examination of four Mycoplasma species in blood of chronic fatigue syndrome patients. FEMS Immunol Med Microbiol. 2002;34(3):209-14.
24. Vernon SD, Shukla SK, Conradt J, et al. Analysis of 16S rRNA gene sequences and circulating cell-free DNA from plasma of chronic fatigue syndrome and non-fatigued subjects. BMC Microbiol. 2002;2:39.
25. Komaroff AL, Goldenberg D. The chronic fatigue syndrome: definition, current studies and lessons for fibromyalgia research. J Rheumatol Suppl. 1989;19:23-7.
26. De Meirleir K, De Becker P, Nijs J, et al. Chronic fatigue syndrome etiology, the immune system and infection. In: Englebienne P, De Meirleir K, editors. Chronic fatigue Syndrome: A biological approach. Boca Raton, Florida: CRC Press; 2002. p. 201-28.
27. Ablin JN, Shoenfeld Y, Buskila D. Fibromyalgia, infection and vaccination: two more parts in the etiological puzzle. J Autoimmun. 2006;27(3):145-52.
28. Nicolson GL, Haier J. Role of chronic bacterial and viral infections in neurodegenerative, neurobehavioral, psychiatric, autoimmune and fatiguing illnesses: Part 2. BJMP. 2010;3(1).
29. Wallace DJ, Hallegua DS. Fibromyalgia: the gastrointestinal link. Curr Pain Headache Rep. 2004;8(5):364-8.
30. Othman M, Aguero R, Lin HC. Alterations in intestinal microbial flora and human disease. Curr Opin Gastroenterol. 2008;24(1):11-6.
31. Akkaya N, Akkaya S, Polat Y, et al. Helicobacter pylori seropositivity in fibromyalgia syndrome. Clinical rheumatology. 2011;30(1):43-9.
32. Buskila D, Shnaider A, Neumann L, et al. Fibromyalgia in hepatitis C virus infection. Another infectious disease relationship. Arch Intern Med. 1997;157(21):2497-500.
33. Rivera J, de Diego A, Trinchet M, et al. Fibromyalgia-associated hepatitis C virus infection. British journal of rheumatology. 1997;36(9):981-5.
34. Kozanoglu E, Canataroglu A, Abayli B, et al. Fibromyalgia syndrome in patients with hepatitis C infection. Rheumatol Int. 2003;23(5):248-51.
35. Narvaez J, Nolla JM, Valverde-Garcia J. Lack of association of fibromyalgia with hepatitis C virus infection. The Journal of rheumatology. 2005;32(6):1118-21.
36. Palazzi C, D'Amico E, D'Angelo S, et al. Hepatitis C virus infection in Italian patients with fibromyalgia. Clinical rheumatology. 2008;27(1):101-3.
37. Adak B, Tekeoglu I, Ediz L, et al. Fibromyalgia frequency in hepatitis B carriers. J Clin Rheumatol. 2005;11(3):157-9.
38. Buskila D, Gladman DD, Langevitz P, et al. Fibromyalgia in human immunodeficiency virus infection. The Journal of rheumatology. 1990;17(9):1202-6.
39. Simms RW, Zerbini CA, Ferrante N, et al. Fibromyalgia syndrome in patients infected with human immunodeficiency virus. The Boston City Hospital Clinical AIDS Team. The American journal of medicine. 1992;92(4):368-74.
40. Cruz BA, Catalan-Soares B, Proietti F. Higher prevalence of fibromyalgia in patients infected with human T cell lymphotropic virus type I. The Journal of rheumatology. 2006;33(11):2300-3.
41. Bennett RM, Jones J, Turk DC, et al. An internet survey of 2,596 people with fibromyalgia. BMC Musculoskelet Disord. 2007;8:27.
42. Buskila D, Neumann L, Vaisberg G, et al. Increased rates of fibromyalgia following cervical spine injury. A controlled study of 161 cases of traumatic injury. Arthritis Rheum. 1997;40(3):446-52.
43. Wynne-Jones G, Jones GT, Wiles NJ, et al. Predicting new onset of widespread pain following a motor vehicle collision. J Rheumatol. 2006;33(5):968-74.
44. Buskila D, Ablin JN, Ben-Zion I, et al. A painful train of events: increased prevalence of fibromyalgia in survivors of a major train crash. Clin Exp Rheumatol. 2009;27(5 Suppl 56):S79-85.
45. Bazzichi L, Rossi A, Giuliano T, et al. Association between thyroid autoimmunity and fibromyalgic disease severity. Clin Rheumatol. 2007;26(12):2115-20.
46. Aarflot T, Bruusgaard D. Association between chronic widespread musculoskeletal complaints and thyroid autoimmunity. Results from a community survey. Scand J Prim Health Care. 1996;14(2):111-5.
47. Pamuk ON, Cakir N. The frequency of thyroid antibodies in fibromyalgia patients and their relationship with symptoms. Clin Rheumatol. 2007;26(1):55-9.
48. Ribeiro LS, Proietti FA. Interrelations between fibromyalgia, thyroid autoantibodies, and depression. J Rheumatol. 2004;31(10):2036-40.
49. Klein R, Bansch M, Berg PA. Clinical relevance of antibodies against serotonin and gangliosides in patients with primary fibromyalgia syndrome. Psychoneuroendocrinology. 1992;17(6):593-8.
50. Loevinger BL, Muller D, Alonso C, et al. Metabolic syndrome in women with chronic pain. Metabolism. 2007;56(1):87-93.
51. Zhou D, Kusnecov AW, Shurin MR, et al. Exposure to physical and psychological stressors elevates plasma interleukin 6: relationship to the activation of hypothalamic-pituitary-adrenal axis. Endocrinology. 1993;133(6):2523-30.
52. De Maio A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: a form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones. 2011;16(3):235-49.
53. Loevinger BL, Shirtcliff EA, Muller D, et al. Delineating psychological and biomedical profiles in a heterogeneous fibromyalgia population using cluster analysis. Clinical rheumatology. 2012;31(4):677-85.
54. Mak TW, Saunders ME. Introduction to the immune response. Chapter 1, Primer to the Immune Response. Academic Cell Update ed. Boston: Elsevier; 2011. p. 3-12.
55. Petersen AM, Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol. 2005;98(4):1154-62.
56. Mak TW, Saunders ME. T cell development activation and effector functions. Chapter 9, Primer to the immune response. Boston: Elsevier; 2011. p. 141-60.
57. Mak TW, Saunders ME. Autoimmune diseases. Chapter 19, Primer to the immune response. Boston: Elsevier; 2011. p. 321-41.
58. Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5(9):675-87.
59. Mak TW, Saunders ME. Immunity to infection. Chapter 13, Primer to the immune response. Boston: Elsevier; 2011. p. 205-26.
60. Aggarwal BB, Sethi G, Nair A, et al. Nuclear factor-kB: A holy grail in cancer prevention and therapy. Current Signal Transduction Therapy. 2006;1:25-52.
61. Bitzer M, von Gersdorff G, Liang D, et al. A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev. 2000;14(2):187-97.
62. Lado-Abeal J, Romero A, Castro-Piedras I, et al. Thyroid hormone receptors are down-regulated in skeletal muscle of patients with non-thyroidal illness syndrome secondary to non-septic shock. Eur J Endocrinol. 2010;163(5):765-73.
63. McKay LI, Cidlowski JA. Cross-talk between nuclear factor-kappa B and the steroid hormone receptors: mechanisms of mutual antagonism. Mol Endocrinol. 1998;12(1):45-56.
64. Brown KD, Claudio E, Siebenlist U. The roles of the classical and alternative nuclear factor-kappaB pathways: potential implications for autoimmunity and rheumatoid arthritis. Arthritis research & therapy. 2008;10(4):212.
65. Gerlo S, Kooijman R, Beck IM, et al. Cyclic AMP: a selective modulator of NF-kappaB action. Cell Mol Life Sci. 2011;68(23):3823-41.
66. Liou JT, Liu FC, Hsin ST, et al. Inhibition of the cyclic adenosine monophosphate pathway attenuates neuropathic pain and reduces phosphorylation of cyclic adenosine monophosphate response element-binding in the spinal cord after partial sciatic nerve ligation in rats. Anesth Analg. 2007;105(6):1830-7.
67. Velazquez KT, Mohammad H, Sweitzer SM. Protein kinase C in pain: involvement of multiple isoforms. Pharmacol Res. 2007;55(6):578-89.
68. Khasar SG, Burkham J, Dina OA, et al. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci. 2008;28(22):5721-30.
69. Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32(12):611-8.
70. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. The New England journal of medicine. 2005;353(16):1711-23.
71. McKay LI, Cidlowski JA. CBP (CREB binding protein) integrates NF-kappaB (nuclear factor-kappaB) and glucocorticoid receptor physical interactions and antagonism. Mol Endocrinol. 2000;14(8):1222-34.
72. Hole JW. The endocrine system. Chapter 13, Human Anatomy and Physiology. Third ed. Dubuque, Iowa: Wm. C. Brown; 1984. p. 433-73.
73. Van Bogaert T, De Bosscher K, Libert C. Crosstalk between TNF and glucocorticoid receptor signaling pathways. Cytokine Growth Factor Rev. 2010;21(4):275-86.
74. Jeay S, Sonenshein GE, Postel-Vinay MC, et al. Growth hormone prevents apoptosis through activation of nuclear factor-kappaB in interleukin-3-dependent Ba/F3 cell line. Molecular endocrinology. 2000;14(5):650-61.
75. Jones KD, Deodhar P, Lorentzen A, et al. Growth hormone perturbations in fibromyalgia: a review. Semin Arthritis Rheum. 2007;36(6):357-79.
76. Cuatrecasas G, Riudavets C, Guell MA, et al. Growth hormone as concomitant treatment in severe fibromyalgia associated with low IGF-1 serum levels. A pilot study. BMC Musculoskelet Disord. 2007;8:119.
77. Jones KD, Burckhardt CS, Deodhar AA, et al. A six-month randomized controlled trial of exercise and pyridostigmine in the treatment of fibromyalgia. Arthritis and rheumatism. 2008;58(2):612-22.
78. Ross RL, Jones KD, Bennett RM, et al. Preliminary Evidence of Increased Pain and Elevated Cytokines in Fibromyalgia Patients with Defective Growth Hormone Response to Exercise. Open Immunol J. 2010;3:9-18.
79. Kawashima S, Hayashi M, Takii T, et al. Serotonin derivative, N-(p-coumaroyl) serotonin, inhibits the production of TNF-alpha, IL-1alpha, IL-1beta, and IL-6 by endotoxin-stimulated human blood monocytes. J Interferon Cytokine Res. 1998;18(6):423-8.
80. Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol. 2011;25(2):285-97.
81. Traynor LM, Thiessen CN, Traynor AP. Pharmacotherapy of fibromyalgia. Am J Health Syst Pharm. 2011;68(14):1307-19.
82. Audhya T, Adams JB, Johansen L. Correlation of serotonin levels in CSF, platelets, plasma, and urine. Biochimica et biophysica acta. 2012;1820(10):1496-501.
83. Nagaya T, Fujieda M, Otsuka G, et al. A potential role of activated NF-kappa B in the pathogenesis of euthyroid sick syndrome. The Journal of clinical investigation. 2000;106(3):393-402.
84. Tapia G, Fernandez V, Varela P, et al. Thyroid hormone-induced oxidative stress triggers nuclear factor-kappaB activation and cytokine gene expression in rat liver. Free Radic Biol Med. 2003;35(3):257-65.
85. Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol. 2000;68(4):437-46.
86. Nicolson GL. Metabolic syndrome and mitochondrial function: molecular replacement and antioxidant supplements to prevent membrane peroxidation and restore mitochondrial function. J Cell Biochem. 2007;100(6):1352-69.
87. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793-801.
88. Zhang F, Basinski MB, Beals JM, et al. Crystal structure of the obese protein leptin-E100. Nature. 1997;387(6629):206-9.
89. Zhang X, Zhang G, Zhang H, et al. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61-73.
90. Tilg H, Moschen AR. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol Metab. 2008;19(10):371-9.
91. Green K, Brand MD, Murphy MP. Prevention of mitochondrial oxidative damage as a therpeutic strategy in diabetes. Diabetes. 2004;53(Suppl 1):S110-S8.
92. Bazzichi L, Ciregia F, Giusti L, et al. Detection of potential markers of primary fibromyalgia syndrome in human saliva. Proteomics Clin Appl. 2009;3(11):1296-304.
93. Silverman MN, Heim CM, Nater UM, et al. Neuroendocrine and immune contributors to fatigue. PM & R : the journal of injury, function, and rehabilitation. 2010;2(5):338-46.
94. Liptan GL. Fascia: A missing link in our understanding of the pathology of fibromyalgia. J Bodyw Mov Ther. 2010;14(1):3-12.
95. Sprott H, Muller A, Heine H. Collagen crosslinks in fibromyalgia. Arthritis and rheumatism. 1997;40(8):1450-4.
96. Ruster M, Franke S, Spath M, et al. Detection of elevated Ne-carboxymethyllysine levels in muscular tissue and in serum of patients with fibromyalgia. Scand J Rheumatol. 2005;34:460-3.
97. Hein G, Franke S. Are advanced glycation end-product-modified proteins of pathogenetic importance in fibromyalgia? Rheumatology (Oxford). 2002;41:1163-7.
98. Bridges D, Thompson SW, Rice AS. Mechanisms of neuropathic pain. Br J Anaesth. 2001;87(1):12-26.
99. De Jongh RF, Vissers KC, Meert TF, et al. The role of interleukin-6 in nociception and pain. Anesth Analg. 2003;96(4):1096-103.
100. Sun J, Ramnath RD, Zhi L, et al. Substance P enhances NF-kappaB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways. Am J Physiol Cell Physiol. 2008;294(6):C1586-96.
101. Martinez-Lavin M, Solano C. Dorsal root ganglia, sodium channels, and fibromyalgia sympathetic pain. Medical hypotheses. 2009;72(1):64-6.
102. Stedman TL. Stedman's medical dictionary. 28th ed. Philadelphia: Lippincott Williams & Wilkins; 2006.
103. Vaeroy H, Helle R, Forre O, et al. Elevated CSF levels of substance P and high incidence of Raynaud phenomenon in patients with fibromyalgia: new features for diagnosis. Pain. 1988;32(1):21-6.
104. Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with the fibromyalgia syndrome. Arthritis and rheumatism. 1994;37(11):1593-601.
105. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res. 2009;15(2):425-30.

The above article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.