



ISSN 1757-8515
Diffuse Alveolar Haemorrhage with ANCA associated vaculitis-review of Literature
Fadi Hammoudeh, Muhammad K. Perwaiz, Setu Patolia, Frances M. Schmidt, Narayan Neupane,Neerja Gulati, Danilo Enriquez, Joseph Quist, Mehjabeen Zahir and Eneh Kennedy
Cite this article as: BJMP 2011;4(1):a402
|
|
Abstract Patients with Wegner’s Granulomatosis often present with diffuse alveolar haemorrhage alongside the classical triad of haemoptysis, anaemia and progressive dyspnoea. The diagnosis is confirmed by bronchoalveolar lavage with serial aspirated aliquots of fluid revealing persistently bloody returns. Lung biopsy is very helpful if it shows granulomatous inflammation and vasculitis however it lacks sensitivity and specificity. Studies suggest that the detection of antineutrophilcytoplasmic antibodies (ANCA) along with Proteinase-3 can substitute for biopsy for the diagnosis of Wegner’s Granulomatosis in patients who present with diffuse alveolar haemorrhage. Keywords: Diffuse Alveolar Haemorrhage (DAH), Wegener’s Granulomatosis (WG), Anti-neutrophil cytoplasmic antibodies (ANCA), classical antineutrophil cytoplasmic antibodies (C-ANCA), anti proteinas-3 (PR3) |
Definition
Diffuse Alveolar Haemorrhage (DAH) is a rare but serious and frequently life-threatening complication of a variety of conditions. DAH refers to a clinical syndrome resulting from injury to the alveolar capillaries, arterioles, and venules leading to red blood cell accumulation in the distal air spaces because of leakage of alveolar capillaries. Most cases of DAH are caused by capillaritis associated with systemic autoimmune diseases such as ANCA-associated vasculitis, anti-GBM disease, and systemic lupus erythematosus.1 Treatment is with immunosuppressants for patients with autoimmune causes and respiratory support if needed.
Diffuse alveolar haemorrhage syndrome is not a specific entity but is a syndrome that suggests a differential diagnosis and a specific sequence of testing.
Aetiology
Many disorders can cause alveolar haemorrhage; they include
- Autoimmune disorders (e.g., systemic vasculitides, Goodpasture's syndrome, antiphospholipid antibody syndrome)
- Pulmonary infections (e.g., invasive aspergillosis, hantavirus infection)
- Toxic exposures (e.g., trimellitic anhydride, isocyanates, crack cocaine, certain pesticides)
- Drug reactions (e.g., propylthiouracil, diphenylhydantoin, amiodarone, methotrexate, , nitrofurantoin, bleomycin, montelukast, infliximab
- Cardiac disorders (e.g., mitral stenosis)
- Coagulation disorders caused by diseases or anticoagulant drugs
- Isolated pauci-immune pulmonary capillaritis
- Idiopathic pulmonary haemosiderosis
- Bone marrow or solid organ transplantation.
Clinical Presentation
The clinical presentation of diffuse alveolar haemorrhage may reflect either alveolar bleeding alone or features of the underlying cause (e.g., haematuria in Wegener granulomatosis, arthritis in systemic lupus erythematosus). Hence, its recognition requires a high degree of suspicion. Some patients present with severe acute respiratory distress requiring mechanical ventilation. However, dyspnoea, cough, and fever are the common initial symptoms and are most often acute or subacute (i.e., present for less than a week). The fever is usually due to the underlying cause, such as lupus. Haemoptysis may be absent at the time of presentation in up to a third of patients because the total alveolar volume is large and can absorb large amounts of blood, without extending more proximally into the airways. Apparent haemoptysis, if present, must be differentiated from haematemesis or pseudohaemoptysis (alveolar flooding with fluid that resembles blood, as in Serratia marcescens pneumonia, in which the reddish hue of the infecting organism can create the impression of alveolar bleeding).
Chest X-ray and Chest CT scan typically shows bilateral infiltrates (figure 1 &2)
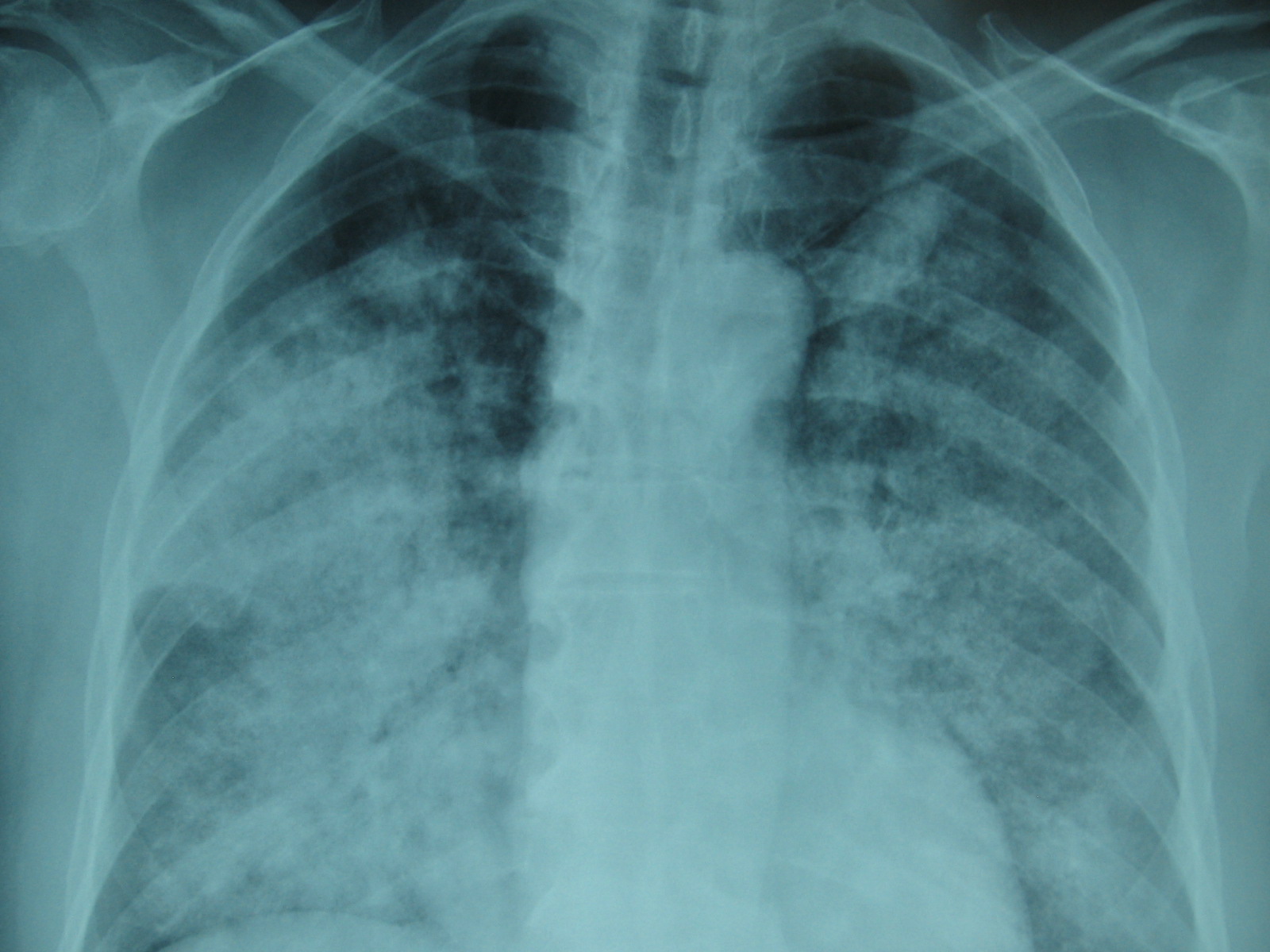
Figure 1
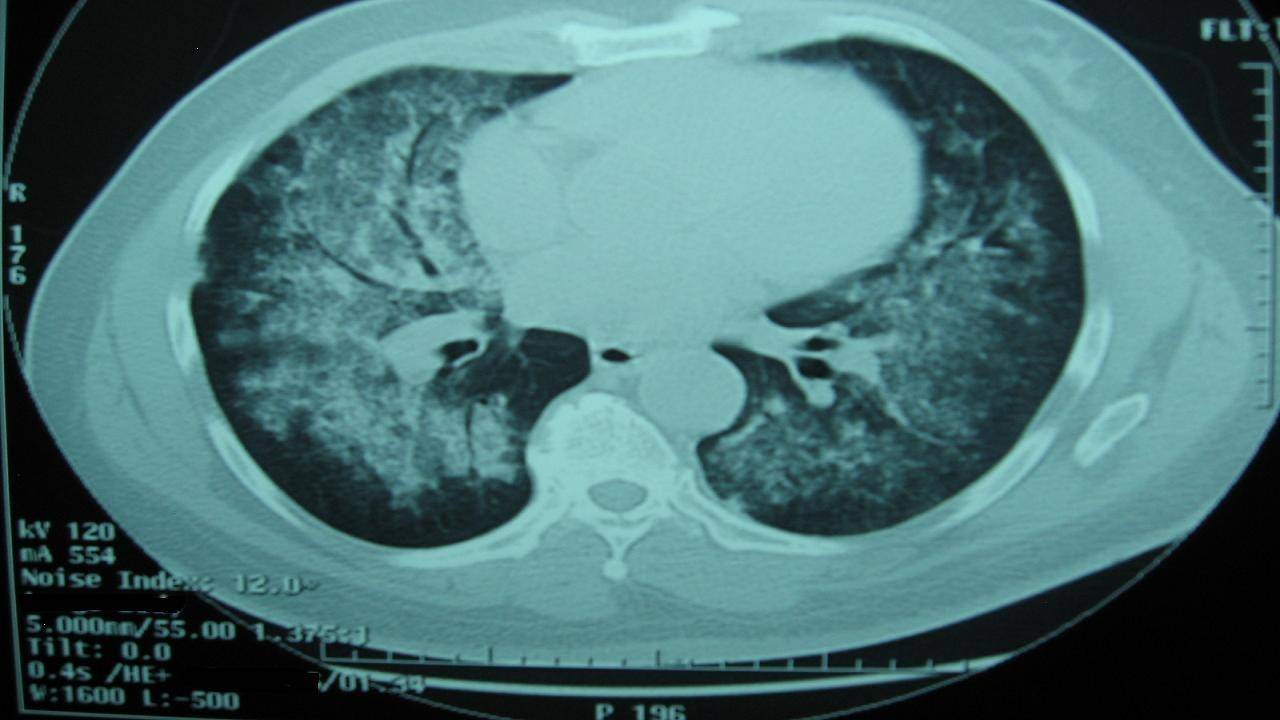
Figure 2
DAH & ANCA associated vasculitides
Wegener's Granulomatosis (WG) is an uncommon disease that affects about 1 in 20,000 to 1 in 30,000 people. WG is defined by the triad of granulomatous inflammation of the respiratory tract, vasculitis of small to medium-size vessels and necrotizing glomerulonephritis. The onset of WG may be indolent with few symptoms, or it may have a rapid and severe onset. About 90% of patients have symptoms of a cold or runny nose or sinusitis that fail to respond to the usual therapeutic measures and last considerably longer than the usual upper respiratory tract infection. Other symptoms include nasal membrane ulcerations and crusting, saddle-nose deformity, inflammation of the ear with hearing problems, inflammation of the eye with sight problems, cough (with or without the presence of blood), pleuritis, (inflammation of the lining of the lung), rash and/or skin sores, fever, lethargy weakness, loss of appetite, weight loss, arthritic joint pain, night sweats, and haematuria which may or may not be indicated by a change in urine colour.Thediagnosis of WG depends on the combination of clinical presentation, serological markers, and histopathological findings. ANCA is a sensitive and specific marker for ANCA-associated systemic vasculitis. In a study done by U. Schönermarck et al,9 624 ANCA- positive patients were included, (C-ANCA: 333, P-ANCA: 291). C-ANCA were highly sensitive (81%) and specific (99.5%) for WG, resulting in high positive predictive value (PPV) (94%). Many studies showed that combining proteinase 3 (PR3) and C-ANCA results(C-ANCA/PR3) increases specificity and Positive Predictive Value close to 100%, but reduces sensitivity close to 70%.10,11,13,14 In summary, the presence of C-ANCA & PR3 antibody is highly suggestive of WG. This led to reevaluation of the role of biopsy for diagnosis of WG in multiple studies.4, 14, 15
The site of biopsy is dependent upon the clinical status. A nasal or sinus biopsy may be the least invasive way to diagnose WG. Renal biopsy is helpful if there is evidence of renal insufficiency or glomerulonephritis. A lung biopsy should only be considered if potentially diagnostic tissue cannot be obtained from any other site.1 Hoffman et al performed a total of 82 open lung biopsies in patients with small vessel vasculitis of which 89% showed evidence of combined vasculitis and necrosis, granulomas and necrosis were found in 90%.16 59 transbronchial biopsies were performed in 48 patients and only four specimens had evidence of vasculitis and granulomas were identified in an additional three. Thus, the role of transbronchial biopsies in these patients is limited and open lung biopsies are more informative but carry a higher morbidity and mortality.
The incidence of DAH has beenreported as between 7-45% in Wegner’s Granulomatosis (WG), and 10-30% in Microscopic Polyangitis (MPA).3, 5, 6 The lungs are the most commonly affected organ in WG with evidence of involvement in over 90% of patients during the course of their disease; in 9% it is the only organ affected. 5,7 In MPA lung involvement is less common than inWG, and occurs in up to 50% of cases during the course of the disease.8 Pulmonary involvement ranges from subclinical changes on high resolution computed tomography to devastating haemoptysis. Approximately 5% of patients will have a fulminant presentation requiring assisted ventilation.
Treatment
Patients with DAH with or without glomerulonephritis, who are found to have ANCA positive can be generally assumed to have WG or MPA. The type of ANCA (PR3-ANCA or MPO-ANCA) found is irrelevant with respect to the initial management of this patients.1 The backbone of therapy is the early identification of disease followed by the rapid induction of disease control with immunosuppression. Early recognition is crucial, because the prompt institution of supportive measures and immunosuppressive therapy is required for survival. The intensity of the initial treatment depends on the severity of the disease. Based on the European Vasculitis Study Group (EUVAS), which categorized the patients in groups according to the severity of their disease, the presence of DAH put the patient in the severe disease group.17 The management of these patients is a combination of corticosteroid and cyclophospamide. S.L Hogan showed that cyclophosphamide reduces mortality and increase the likelihood of inducing remission in patients with ANCA-associated vasculitis. 18
DAH is animportant cause of morbidity and mortality in ANCA- associated vasculitis, the mortality rate may reach 66%, which is six times greater than vasculitis without alveolar hemorrhage.3,19,20,21 Based on the high mortality rate with DAH in ANCA-associated vasculitis, and reduction in mortality shown with cyclophosphamide, treatment with cyclophosphamide should be started as early as possible, based on the clinical presentation and the presence of ANCA, without waiting histological confirmation.
Conclusion
DAH leading to acute respiratory distress syndrome is a rare and life threatening condition in adults with ANCA positive vasculitis. Patients with DAH with or without glomerulonephritis, who are found to have ANCA positive can be generally assumed to have WG or MPA, and diagnostic lung biopsy may be deferred. Early institution of treatment with prednisone and cyclophosphamide can significantly reduce morbidity and mortality.
|
Key points
1. Patients with Wegner’s Granulomatosis often present with diffuse alveolar haemorrhage. These patients must be treated promptly as delay in treatment results in high morbidity and mortality.
2. Lung biopsy is very helpful if it shows granulomatous inflammation and vasculitis however it lacks sensitivity and specificity.
3. Detection of C-ANCA with Proteinase-3 can substitute for biopsy in the diagnosis of WG in patients who present with diffuse alveolar haemorrhage.
|
|
Competing Interests None declared Author Details <p> Fadi Hammoudeh MD, Muhammad K. Perwaiz MD, Setu Patolia MD, Frances M. Schmidt MD, Narayan Neupane, MD, Neerja Gulati MD, Danilo Enriquez MD, Joseph Quist,MD Mehjabeen Zahir MD, Eneh Kennedy MD, - Interfaith Medical Center at 1545 Atlantic Avenue Brooklyn, NY</p> CORRESPONDENCE: Muhammad K. Perwaiz MD, Fellow pulmonary department, Interfaith medical center at 1545 Atlantic Avenue Brooklyn, NY Email: fhammoudeh@interfaithmedical.com |
References
1. Specks U. Diffuse alveolar haemorrhage syndromes. Curr Opin Rheumatol 2001; 13:12-17.2. Travis W. Colby T. Lombard C, et al: A clinicopathologic study of 34 cases of diffuse pulmonary haemorrhage with lung biopsy confirmation. Am J Surg Pathol 1990 ;14:11123. D. R. Thickett, A. G. Richter, N. Nathani, G. D. Perkins and L. HarperPulmonary manifestations of anti-neutrophil cytoplasmic antibody (ANCA)-positive vasculitis. Rheumatology 2006;45:261–2684. Travis WD, Hoffman GS, Leavitt RY et al. Surgical pathology of the lung in Wegener's granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol. 1991;15(4):315-335. J F Cordier, D Valeyre, L Guillevin, R Loire and J M Brechot Pulmonary Wegener's granulomatosis. A clinical and imaging study of 77 cases. Chest 1990; 97: 906-9126. S J HAWORTH, C 0 S SAVAGE, D CARR, J M B HUGHES, A J REES Pulmonary haemorrhage complicating Wegener's granulomatosis and microscopic polyarteritis British Medical Journal. 1985;290(15);1775-17787. Aine Burns Pulmonary Vasculitis Thorax 1998; 53:220–2278. Octavian C. Ioachimescu. Diffuse alveolar haemorrhage: Diagnosing it and finding the cause. Cleveland Clinic Journal of Medicine .2008;75(4): 258-2809. U. Schönermarck, P. Lamprecht, E. Csernok, W. L. Gross. Prevalence and spectrum of rheumatic diseases associated with proteinase 3-antineutrophil cytoplasmic antibodies (ANCA) and myeloperoxidase-ANCA. Rheumatology 2001;40:178-18410. Langford CA. Wegener granulomatosis. Am J Med Sci 2001;321:76-82.11. Falk RJ, Jennette JC. ANCA small-vessel vasculitis. J Am Soc Nephrol 1997; 8:314-22.12. Hagen EC, Daha MR, Hermans J et al. Diagnostic value of standardized assays for anti neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization Kidney Int. 1998;53(3):743–53.13. Moosig F, Lamprecht P, Gross WL. Wegener's Granulomatosis: the current view. Clin Rev Allergy Immunol. 2008;35(1-2):19-2114. Bosch X, Guilabert A, Espinosa G, et al. Treatment of antineutrophil cytoplasmic antibody associated vasculitis: a systematic review. JAMA. 2007; 298(6):655–6915. Mar EJ, Matsubara O, Nelia S. Tan-Liu et al. The pulmonary biopsy in the early diagnosis of Wegener's (pathergic) granulomatosis: A study based on 35 open lung biopsies. Hum Pathol. 1988;19(9):1065-7116. Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener’s granulomatosis: an analysis of 158 patients. Ann InternMed 1992;116:488–9817. Stephen K. Frankel, Gregory P. Cosgrove, Aryeh Fischer, Richard T. Meehan and Kevin K. Brown Update in the Diagnosis and Management of Pulmonary Vasculitis Chest 2006;129;452-46518. SL Hogan, PH Nachman, AS Wilkman, JC Jennette and RJ Falk Prognostic markers in patients with antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis Journal of the American Society of Nephrology.1996;7:23-3219. Gisele Zandman-Goddard MD Diffuse Alveolar Haemorrhage in Autoimmune Diseases. IMAJ 2002;4:461-46220. Lin Y, Zheng W, Tian X, Zhang X, Zhang F, Dong Y. Antineutrophil cytoplasmic antibody-associated vasculitis complicated with diffuse alveolar haemorrhage: a study of 12 cases. J Clin Rheumatol. 2009;15(7):341-4.21. Chen GX, Dong Y, Ju ZB . A clinical analysis of 32 patients with diffuse alveolar haemorrhage in diffuse connective tissue diseases. Zhonghua Nei Ke Za Zhi. 2008;47(5):362-5

The above article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.