Diffuse Alveolar Haemorrhage (DAH) is a rare but serious and frequently life-threatening complication of a variety of conditions. DAH refers to a clinical syndrome resulting from injury to the alveolar capillaries, arterioles, and venules leading to red blood cell accumulation in the distal air spaces because of leakage of alveolar capillaries. Most cases of DAH are caused by capillaritis associated with systemic autoimmune diseases such as ANCA-associated vasculitis, anti-GBM disease, and systemic lupus erythematosus.1 Treatment is with immunosuppressants for patients with autoimmune causes and respiratory support if needed.
Diffuse alveolar haemorrhage syndrome is not a specific entity but is a syndrome that suggests a differential diagnosis and a specific sequence of testing.
Aetiology
Many disorders can cause alveolar haemorrhage; they include
Coagulation disorders caused by diseases or anticoagulant drugs
Isolated pauci-immune pulmonary capillaritis
Idiopathic pulmonary haemosiderosis
Bone marrow or solid organ transplantation.
Clinical Presentation
The clinical presentation of diffuse alveolar haemorrhage may reflect either alveolar bleeding alone or features of the underlying cause (e.g., haematuria in Wegener granulomatosis, arthritis in systemic lupus erythematosus). Hence, its recognition requires a high degree of suspicion. Some patients present with severe acute respiratory distress requiring mechanical ventilation. However, dyspnoea, cough, and fever are the common initial symptoms and are most often acute or subacute (i.e., present for less than a week). The fever is usually due to the underlying cause, such as lupus. Haemoptysis may be absent at the time of presentation in up to a third of patients because the total alveolar volume is large and can absorb large amounts of blood, without extending more proximally into the airways. Apparent haemoptysis, if present, must be differentiated from haematemesis or pseudohaemoptysis (alveolar flooding with fluid that resembles blood, as in Serratia marcescens pneumonia, in which the reddish hue of the infecting organism can create the impression of alveolar bleeding).
Chest X-ray and Chest CT scan typically shows bilateral infiltrates (figure 1 &2)
Figure 1
Figure 2
DAH & ANCA associated vasculitides
Wegener's Granulomatosis (WG) is an uncommon disease that affects about 1 in 20,000 to 1 in 30,000 people. WG is defined by the triad of granulomatous inflammation of the respiratory tract, vasculitis of small to medium-size vessels and necrotizing glomerulonephritis. The onset of WG may be indolent with few symptoms, or it may have a rapid and severe onset. About 90% of patients have symptoms of a cold or runny nose or sinusitis that fail to respond to the usual therapeutic measures and last considerably longer than the usual upper respiratory tract infection. Other symptoms include nasal membrane ulcerations and crusting, saddle-nose deformity, inflammation of the ear with hearing problems, inflammation of the eye with sight problems, cough (with or without the presence of blood), pleuritis, (inflammation of the lining of the lung), rash and/or skin sores, fever, lethargy weakness, loss of appetite, weight loss, arthritic joint pain, night sweats, and haematuria which may or may not be indicated by a change in urine colour.Thediagnosis of WG depends on the combination of clinical presentation, serological markers, and histopathological findings. ANCA is a sensitive and specific marker for ANCA-associated systemic vasculitis. In a study done by U. Schönermarck et al,9 624 ANCA- positive patients were included, (C-ANCA: 333, P-ANCA: 291). C-ANCA were highly sensitive (81%) and specific (99.5%) for WG, resulting in high positive predictive value (PPV) (94%). Many studies showed that combining proteinase 3 (PR3) and C-ANCA results(C-ANCA/PR3) increases specificity and Positive Predictive Value close to 100%, but reduces sensitivity close to 70%.10,11,13,14 In summary, the presence of C-ANCA & PR3 antibody is highly suggestive of WG. This led to reevaluation of the role of biopsy for diagnosis of WG in multiple studies.4, 14, 15
The site of biopsy is dependent upon the clinical status. A nasal or sinus biopsy may be the least invasive way to diagnose WG. Renal biopsy is helpful if there is evidence of renal insufficiency or glomerulonephritis. A lung biopsy should only be considered if potentially diagnostic tissue cannot be obtained from any other site.1 Hoffman et al performed a total of 82 open lung biopsies in patients with small vessel vasculitis of which 89% showed evidence of combined vasculitis and necrosis, granulomas and necrosis were found in 90%.16 59 transbronchial biopsies were performed in 48 patients and only four specimens had evidence of vasculitis and granulomas were identified in an additional three. Thus, the role of transbronchial biopsies in these patients is limited and open lung biopsies are more informative but carry a higher morbidity and mortality.
The incidence of DAH has beenreported as between 7-45% in Wegner’s Granulomatosis (WG), and 10-30% in Microscopic Polyangitis (MPA).3, 5, 6 The lungs are the most commonly affected organ in WG with evidence of involvement in over 90% of patients during the course of their disease; in 9% it is the only organ affected. 5,7 In MPA lung involvement is less common than inWG, and occurs in up to 50% of cases during the course of the disease.8 Pulmonary involvement ranges from subclinical changes on high resolution computed tomography to devastating haemoptysis. Approximately 5% of patients will have a fulminant presentation requiring assisted ventilation.
Treatment
Patients with DAH with or without glomerulonephritis, who are found to have ANCA positive can be generally assumed to have WG or MPA. The type of ANCA (PR3-ANCA or MPO-ANCA) found is irrelevant with respect to the initial management of this patients.1 The backbone of therapy is the early identification of disease followed by the rapid induction of disease control with immunosuppression. Early recognition is crucial, because the prompt institution of supportive measures and immunosuppressive therapy is required for survival. The intensity of the initial treatment depends on the severity of the disease. Based on the European Vasculitis Study Group (EUVAS), which categorized the patients in groups according to the severity of their disease, the presence of DAH put the patient in the severe disease group.17 The management of these patients is a combination of corticosteroid and cyclophospamide. S.L Hogan showed that cyclophosphamide reduces mortality and increase the likelihood of inducing remission in patients with ANCA-associated vasculitis. 18
DAH is animportant cause of morbidity and mortality in ANCA- associated vasculitis, the mortality rate may reach 66%, which is six times greater than vasculitis without alveolar hemorrhage.3,19,20,21 Based on the high mortality rate with DAH in ANCA-associated vasculitis, and reduction in mortality shown with cyclophosphamide, treatment with cyclophosphamide should be started as early as possible, based on the clinical presentation and the presence of ANCA, without waiting histological confirmation.
Conclusion
DAH leading to acute respiratory distress syndrome is a rare and life threatening condition in adults with ANCA positive vasculitis. Patients with DAH with or without glomerulonephritis, who are found to have ANCA positive can be generally assumed to have WG or MPA, and diagnostic lung biopsy may be deferred. Early institution of treatment with prednisone and cyclophosphamide can significantly reduce morbidity and mortality.
Key points
1.Patients with Wegner’s Granulomatosis often present with diffuse alveolar haemorrhage. These patients must be treated promptly as delay in treatment results in high morbidity and mortality.
2.Lung biopsy is very helpful if it shows granulomatous inflammation and vasculitis however it lacks sensitivity and specificity.
3.Detection of C-ANCA with Proteinase-3 can substitute for biopsy in the diagnosis of WG in patients who present with diffuse alveolar haemorrhage.
Currently, peripheral vascular disease (PVD), causing an inadequate oxygen supply to the limbs, globally affects no less than 3–10% of the population1. Peripheral vascular disease, including diabetic foot, arteriosclerosis obliterans, and thromboangitis obliterans, commonly affect the arteries supplying the leg. Based on the severity of the symptoms, usually two clinical presentations are distinguished: intermittent claudication (IC) is characterised by pain upon walking while critical limb ischaemia (CLI) is a more severe form in which pain occurs at rest and which is accompanied by necrosis and ulceration.
Peripheral arterial occlusive disease (PAOD) is estimated to develop in 500 to 1000 individuals per million persons per year2, 3. The prevalence of all stages of PAOD in the general population is estimated to be 4.2% to 35%. Within this group, 4.3% to 9.6% will experience progression of the disease towards CLI, eventually resulting in amputation of the affected limb4. Diabetic PAOD patients are at the highest risk within this patient group: they are about 10 times more likely to come to amputation, and the prevalence of gangrene is 20 to 30 times higher2. CLI has important functional implications and a major impact on the quality of life. Quality of life indices of patients with CLI have been reported to be similar to those of terminal cancer patients5. In addition, CLI is associated with surgery and hospitalisation6. CLI is also associated with increased mortality (the 1-year mortality is approximately 25% and may be as high as 45% after amputation)7 , and even asymptomatic PAOD by itself is a significant predictor of cardiovascular morbidity and death8. While obstructive atherosclerotic disease is the most common cause of PVD, some forms of vasculitis, such as thromboangiitis obliterans or Buerger’s disease, also result in peripheral ischaemia (in feet and/or hands), often progressing to tissue loss and major amputations9,10.
Unfortunately, a significant proportion of patients (including both IC and CLI cases) are not eligible for or do not beneficially respond to these revascularisation procedures due to the widespread nature or the distal location of the obstructions or due to the presence of co-morbidities putting them at higher risk for peri-procedural death.
For these ‘no-option’ patients, non-invasive revascularisation strategies have been introduced, which fall into two categories: single gene/protein-based or cell-based strategies. Angiogenic growth factor (e.g., vascular endothelial growth factor (VEGF), fibroblast growth factors (FGFs), and hepatocyte growth factor) therapy has been tested clinically since more than 5 years. But the overall benefit for PVD patients has been disappointing11.
Consequently, exploring new strategies for revascularisation of ischaemic limbs is of major importance.
What are stem cells?
Stem cells are defined as a cell population capable of self-renewal, proliferation and differentiation. They serve as a repair system for the body.
Stem cells are classified into two different types during the development of the organism: embryonic stem cells and adult stem cells (ASCs).
The use of adult stem cells in research and therapy is not as controversial as embryonic stem cells, because the production of ASC does not require the destruction of an embryo. Additionally, because in some instances ASC can be obtained from the intended recipient, (an autograft) the risk of rejection is essentially non-existent in these situations.
Where are adult stem cells found, and what do they normally do?
Adult stem cells (ASCs) have been identified in many organs and tissues, including brain, bone marrow, peripheral blood, blood vessels, skeletal muscle, skin, teeth, heart, gut, liver, ovarian epithelium, and testis. In HYPERLINK "http://en.wikipedia.org/wiki/Adult" \o "Adult"adult organisms, stem cells and HYPERLINK "http://en.wikipedia.org/wiki/Progenitor_cell" \o "Progenitor cell"progenitor cells act as a repair system for the body, replenishing specialised cells, but also maintain the normal turnover of regenerative organs, such as blood, skin or intestinal tissues. They are thought to reside in a specific area of each tissue (called a "stem cell niche"). In many tissues, current evidence suggests that some types of stem cells are pericytes, cells that compose the outermost layer of small blood vessels. Stem cells may remain quiescent (non-dividing) for long periods of time until they are activated by a normal need for more cells to maintain tissues, or by disease or tissue injury.
The concept of stem cell based revascularisation emerged in 1997, when Isner’s group described circulating cells in adults called endothelial progenitor cells (EPC) with the capacity to differentiate into endothelial cells (EC) and incorporate into new vessels in ischaemic tissue12. Since then, the number of studies reporting on stem cell related revascularisation has exponentially increased. Bone marrow (BM) derived stem cells have been identified as a potential new therapeutic target. Most adult stem cells are lineage-restricted (multipotent) and are generally referred to by their tissue origin for example: mesenchymal stem cell, adipose-derived stem cell, endothelial stem cell etc13, 14.
In the 1950s, researchers discovered that the bone marrow contains at least two kinds of stem cells. One population, called hematopoietic stem cells, forms all the different types of blood cells in the body. A second population, called bone marrow stromal stem cells (also called mesenchymal stem cells, or skeletal stem cells by some), were discovered a few years later. These non-hematopoietic stem cells make up a small proportion of the stromal cell population in the bone marrow, and can generate bone, cartilage, fat, cells that support the formation of blood, and fibrous connective tissue.
Adult stem cell treatments have been successfully used for many years to treat leukaemia and related bone/blood cancers through bone marrow transplant15.
Relationship between neoangiogenesis and cell population.
Neoangiogenesis:
Three concepts of vascular growth have been described to date—angiogenesis, vasculogenesis, and arteriogenesis (collateral artery growth)—which represent different aspects of an integrated process. Stimulation of arteriogenesis seems clinically most relevant and has most recently been attempted using autologous bone marrow transplantation with some beneficial results, although the mechanism of action is not completely understood.
Cell population:
Hematopoietic stem cells may be CD34+ AC133+ or CD34- AC133+ or CD34+ AC133-. Vascular development is regulated by growth factors and their receptors such as vascular endothelial growth factor (VEGF) and VEGF tyrosine kinase receptors such as VEGFR-1 (flt-1) or VEGFR-2 (KDR or flk-1). Other growth factors such as angiopoietin-1 that bind a tyrosine kinase receptor Tie-2 may be involved in completing the vascular architecture by assembling pericytes and smooth muscle cells around endothelial cells16.
Marrow or peripheral blood CD34+ hematopoietic stem cells express VEGFR and Tie.12 When cultured ex-vivo in fibronectin-coated flasks with VEGF, CD34+ AC133+ cells differentiate into endothelial cells by morphology, acetylated low-density lipoprotein incorporation, nitric oxide release, Von Willebrand factor expression, and lectin binding17.
The unfractionated mixture of hematopoietic mononuclear cells includes more differentiated cells that are thought to provide angiogenic cytokines as well as stem cells that become incorporated into collateral vessels by a process of neoangiogenesis. In clinical trials, Tateishi-Yuyama et al.18 injected autologous bone marrow mononuclear cells into patients with ischaemic PVD. Patients were selected for chronic ischaemic extremity pain or non-healing ischaemic ulcers or both and a resting blood pressure ankle-brachial index less than 0.6. Bone marrow cells were collected under general anaesthesia from the posterior superior iliac crest and with a 26-gauge needle injected into the gastrocnemius muscle of the ischaemic leg in multiple sites divided by a 3x3 cm grid. Significant improvement in the ABI, trans-cutaneous oxygen pressure, and pain-free walking occurred following treatment18.
Several independent clinical studies have reported beneficial effects of the administration of bone marrow mononuclear cells (BM-MNC), Granulocyte Colony Stimulating Factor (G-CSF) mobilised Peripheral Blood Mononuclear Cells (PB-MNC), G-CSF-mobilised PB-MNC after ex vivo culturing, G-CSF mobilised CD34+ cells, and G-CSF mobilised CD133+ cells in patients with CLI. However, no direct comparisons have been performed and it is still unclear which cell types or subpopulations provide the best treatment results. The progenitor cells specifically involved in vascular repair and neovascularisation were initially thought to originate from the CD34+ hematopoietic progenitor cell population, analogous to the common hemangioblast precursor in embryonic development19, 20.
Consistently, in the Therapeutic Angiogenesis using Cell Transplantation (TACT) study, legs that were injected with PB-MNC, containing approximately 500-fold less CD34+ cells than BM-MNC, showed much smaller increases in collateral perfusion as compared with BM-MNC-injected legs.18,21 Furthermore, Saigawa et al demonstrated a correlation between the number of implanted CD34+ cells and the efficacy of bone marrow implantation21.
However, several studies suggest that CD34- cell populations also play an important role in the beneficial effects of BM cell therapy. Asahara et al already showed that CD34- cells, added to CD34+ cells in culture, improved outgrowth of cells with an endothelial phenotype12. Co-culture of CD34+ cells with CD34-cells in an in-vitro 3-D matrix model using human microvascular endothelial cells significantly enhanced neovascularisation as compared with CD34+ cells alone22.Other groups described that non-hematopoietic bone marrow mesenchymal precursor cells and myeloid/monocyte lineage cells (CD14+) can also differentiate into EPC or into cells with EPC characteristics23-26. Iba et al compared the angiogenic effects of the same numbers of BM-MNC and PB-MNC (containing 2.4% and 0.02% CD34+ cells, respectively) in a rat hind limb ischaemia model and showed that although there was no incorporation of PB-MNC, the angiogenic effect of PB-MNC was approximately 72% relative to that of BM-MNC27. Moreover, Tateno et al showed that there was no significant difference in stimulation of neovascularisation after infusion of PB-MNC and BM-MNC10.
These data suggest that, apart from incorporation of EPC, EPC supply of angiogenic factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor, and angiopoietin-1 plays an important role. This role of the paracrine effects of EPC on vascular growth have also been demonstrated by the group of Schaper28, 29.
A recent report proposed that implanted cells stimulate muscle cells to produce angiogenic factors, thereby promoting neovascularisation10. Yang and co-workers reported a simple and effective therapeutic approach for diabetic limb ischaemia by autologous transplantation of G-CSF -mobilised peripheral blood stem cells30.
Thus, different cell populations are involved in vascular repair and neovascularisation, and these cells may act via direct incorporation into the endothelial layer and endothelial differentiation, by supply of angiogenic factors, or by a combination of both31.
The majority of studies on cell therapy for CLI have used whole MNC fractions and at this moment it is unclear whether administration of more selected cell populations or ex-vivo culture toward an endothelial phenotype would be more effective.
Although clinical studies showed promising results from both BM-MNC and G-CSF-mobilized PB-MNC, recent data suggest that functional activity of the G-CSF mobilised cells, as assessed by the migratory response to VEGF and stromal cell-derived factor1, is significantly reduced as compared with non-mobilised cells from the same patient. Also in in-vivo experiments in nude mice with hind limb ischaemia, G-CSF-mobilised EPC show a reduced capacity to augment blood flow recovery and to prevent necrosis as compared with the same EPC without G-CSF stimulation32.
It is important to note that cell isolation protocols may also have a major impact on the functional activity of BM-derived progenitor cells33.
Optimal Dosage
It is remarkable that all studies discussed above reportfavourable outcome, despite varying dosages, with an even so varying concentration of CD34+ cells. In the studies involving BM cell administration, amounts of aspiratedBM cell ranging from 80 to 1000 ml, from which theinjected dosage of progenitor cells was retrieved, were reported.In the TACT Study18 and in the study by Higashi etal.34 approximately 1.6x 109 MNC were obtained from 500ml of BM, whereas Durdu et al.9 retrieved a 50-fold of MNCfrom the same amount of BM (101x109 MNC from 653 mlof BM). Bartsch et al.35 separated a 2.5 times smaller amountof MNC from the same amount of BM (0.1x109 MNC from80 ml of BM). The fraction of CD34+ cells in the isolatedMNC population varies from 0.6% in the study by Kajiguchiet al.36 to 2.4% in the TACT study18.
Clinical Evaluation
Currently used measures for clinical evaluation, such as ankle-brachial pressure index, are subject to factors other than improvements in perfusion alone. In accordance with the Trans-Atlantic Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC-II) recommendations, future trials should ideally combine multiple measures for clinical improvement and quantification of the arterial flow to evaluate treatment success, which include ankle pressure, toe pressures, TcPO2, microcirculation investigation methods like laser Doppler fluxometry, and anatomic imaging1.
In addition, questionnaires addressing pain experience, pain “magnitude” (pain intensity, emotion, cognitive-evaluative, and sensitivity) and pain at rest (on a visual analogue scale), as well as quality of life questionnaires will provide patient-based parameters for the clinical effects of therapy.
Ulcer status should be assessed by measurement of the cumulative total ulcer area, with ulcer healing defined as healing of all ulcers of the treated leg. Limb status can be assessed using the criteria of Rutherford37.
Contrast-enhanced high spatial resolution magnetic resonance angiography is a reproducible and robust modality for assessment and quantification of new vessel formation, detecting different sizes of collateral vessels, and determination of (changes in) tissue perfusion.
However, Choksy and Chan38 pointed out that a major scientific weakness in angiogenesis research lies in the assessment of vascular growth.
Avenues to explore?
How do adult stem cells evolve during development and how are they maintained in the adult? Are they "leftover" embryonic stem cells, or do they arise in some other way?
If the beneficial effect of adult stem cell transplantation is a trophic effect, what are the mechanisms?
What are the factors that control adult stem cell proliferation and differentiation?
What are the factors that stimulate stem cells to relocate to sites of injury or damage, and how can this process be enhanced for better healing in PVD?
Why do stem cells remain in an undifferentiated state when all the cells around them have differentiated? What are the characteristics of their “niche” that controls their behaviour?
How can assessment of neo-angiogenesis be improved?
Conclusion
Clearly, stem cell safety must be scrutinised and assessed throughout the entire treatment or research process. Guidelines and strategies must also be developed to ensure that every aspect of stem cell use - from identification and isolation of stem cells to stem cell transplant - is stringently coordinated.
Although several clinical studies show promising results, larger randomised, blinded, placebo-controlled trials are needed to provide definite proof of the clinical effects of adult stem cell therapy in these patients. In addition, questions regarding the cell population(s) to be used, optimal dose, and routes of administration will have to be addressed. If doctors and scientists can establish safe protocols for stem cell use, everyone can benefit from the full potential of the remarkable and possibly life-saving stem cell therapies.
Irritable bowel syndrome (IBS) is a common disorder characterized by abdominal pain and altered bowel habit for at least three months.(1)
IBS is further defined depending on the predominant bowel symptom: IBS with constipation (IBS-C) or IBS with diarrhoea (IBS-D). Those not classified as either IBS-C or IBS-D are considered as mixed IBS (IBS-M). Alternating IBS (IBS-A) defines patients whose bowel habits oscillate from diarrhoea to constipation and vice versa.
IBS is a prevalent and expensive condition that is associated with a significantly impaired health-related quality of life (HRQOL) and reduced work productivity. IBS care consumes over $ 20 billion in both direct and indirect expenditures. Moreover, patients with IBS consume over 50% more health care resources than matched controls without IBS.(1)Based on strict criteria, 7 – 10 % of people have IBS worldwide. Community-based data indicate that diarrhoea-predominant IBS (IBS-D) and mixed IBS (IBS-M) subtypes are more prevalent than constipation-predominant IBS (IBS-C), and that switching among subtype groups may occur. IBS is 1.5 times more common in women than in men, is more common in lower socioeconomic groups, and is more commonly diagnosed
in patients younger than 50 years of age. Prevalence estimates of IBS range from 1 % to more than 20% in North America(7%).(1)In Asia the prevalence is about 5%.(3,4,5)Recently, a School-Based Study in chinareportedthe prevalence of IBS in adolescents and children was 13.25% and the ratio of boys to girls was 1:1.8.(6)Most patient with IBS in India are middle-aged men (mean age 39.4 years).(7)
Underlying pathophysiology:
Given the lack of definitive organic markers for IBS, the absence of aconsolidatedhypothesis regarding its underlying pathophysiology is not surprising. Nevertheless, important advances in research made during the past 50 years have brought us closer than ever to understanding the numerous existing aetiological factors involved in this multifaceted disorder, including environmental factors, genetic factors, previous infection, food intolerance, and abnormal serotonergic signaling in the GI tract.
Environmental factors:
The biopsychosocial model proposed by Engel(8)takes into account the interplay between biologic, psychological, and social factors. This model proposes that there is an underlying biologic predisposition for IBS that may be acted on by environmental factors and psychological stressors, which contribute to disease development, the patient's perception of illness, and impact on treatment outcomes. Different studies have shown that stress can result in release of stress-related hormones that affect colonic sensorimotor function (eg, corticotropin-releasing factor [CRF] and inflammatory mediators [eg, interleukin (IL)-1]), leading to inflammation and altering GI motility and sensation.
Genetics factors :
Twin studies have shown that IBS is twice as prevalent in monozygotic twins than in dizygotic twins.(9,10,11)IBS may be associated with selected gene polymorphisms, including those in IL-10, G-protein GNb3, alpha adrenoceptor, and serotonin reuptake transporter (SERT).
Post-infectious IBS (PI-IBS):
Culture positive gastroenteritis is a very strong risk factor for IBS. Different prospective studies show IBS symptoms developed in 7% to 32% of patients after they recovered from bacterial gastroenteritis.(12,13,14)Specific risk factors for the development of PI-IBS have been identified, including younger age, female sex, presence of severe infectious gastroenteritis for a prolonged period, use of antibiotics to treat this infection, and presence of concomitant psychological disorders (eg, anxiety).(12,13,15,16)
Small Intestinal bacterial overgrowth
Pimentel and colleagues(17,18)have shown that, when measured by the lactose hydrogen breath test (LHBT), small intestinal bacterial overgrowth (SIBO) has been detected in 78% to 84% of patients with IBS. Hence, a higher than usual population of bacteria in the small intestine has been proposed as a potential aetiological factor in IBS. While another study involving a review for the presence of gastrointestinal-related symptoms (including IBS) has shown that asensitivity of the LHBT for SIBO has been shown to be as low as 16.7%, and specificity approximately 70% and the test alone for small intestinal bacterial overgrowth were poor. Hence, combination with scintigraphy resulted in 100% specificity to assess the treatment responce, because double peaks in serial breath hydrogen concentrations may occur as a result of lactulose fermentation by cecal bacteria. (19,20)
Food intolerance :
Approximately 60% of IBS patients believe and different studies show that allergy to certain foods could trigger IBS symptoms. Recent research involving exclusion of foods patients had immunoglobulin (Ig) G antibodies, which are associated with a more delayed response after antigen exposure than IgE antibodies, resulted in significantly better symptom improvement than in patients in the non-exclusion group.(21)
Serotonin signaling in Gastrointestinal (GI) tract:
Normal gut physiology is predicated to be an interaction between the GI musculature and the autonomic nervous system (ANS), and central nervous system (CNS) by the neurotransmitter serotonin (5-hydroxytryptamine [5-HT]) . Impairment in this interaction affects GI motility, secretion, and visceral sensitivity leading to the symptoms associated with IBS .(22)
Preliminary steps toward making a positive diagnosis of IBS:
A careful history and physical examination are frequently helpful in establishing the diagnosis. A variety of criteria have been developed to identify a combination of symptoms to diagnose IBS. Different guidelines from different studies help in making a positive diagnosis of IBS based primarily on the pattern and nature of symptoms, without the need for excessive laboratory testing. In 1978, Manning and colleagues(23,24) proposed diagnostic criteria for IBS that were found to have a reasonable sensitivity of 78% and a specificity of 72%.(1)In 1984, Kruis and colleagues developed another diagnostic criteria with a high sensitivity of 77% and a specificity 89%. Likewise, in 1990 Rome I(25)criteria came with a sensitivity of 71% and specificity of 85%. RomeII(1999)(26)and Rome III(2006)(27)have not been evaluated yet. None of the symptom based diagnostic criteria have been evaluated and ideal reliability found.(1)
Summary of diagnostic criteria used to define IBS:(1)
In 1978, Manning defined IBS as a collection of symptoms, given below, but did not describe their duration. The number of symptoms that need to be present to diagnose IBS was also not reported in the paper, but a threshold of three positive is the most commonly used:
a) Abdominal pain relieved by defecation
b) More frequent stools with onset of pain
c) Looser stools with onset of pain
d) Mucus per rectum
e) Feeling of incomplete emptying
f) Patient-reported visible abdominal distension
Kruis in 1984, defined IBS by a logistic regression model that describes the probability of IBS. Symptoms need to be present for more than two years. Symptoms are as follows:
a) Abdominal pain, flatulence, or bowel irregularity
b) Description of character and severity of abdominal pain
c) Alternating constipation and diarrhea
Signs that exclude IBS (each determined by the physician) :
a) Abnormal physical findings and/or history pathognomonic for any diagnosis other than IBS
b) Erythrocyte sedimentation rate >20 mm/2 h
c) Leukocytosis >10,000/cc
d) Anaemia (Hemoglobin < 12 for women or < 14 for men)
e) Impression, the physician could perform a PR and see blood or the patient may report it.
Again in 1990, Rome I defined IBS as abdominal pain or discomfort relieved with defecation, or associated with a change in stool frequency or consistency, PLUS two or more of the following symptoms on at least 25% of occasions or days for three months:
a) Altered stool frequency
b) Altered stool form
c) Altered stool passage
d) Passage of mucus
e) Bloating or distension
Rome II, in 1999, redefined the criteria as abdominal discomfort or pain that has two of three features for 12 weeks (need not be consecutive) in the last one year.
a) Relieved with defecation
b) Onset associated with a change in frequency of stool
c) Onset associated with a change in form of stool
Recently , Rome III (2006) defined IBS as recurrent abdominal pain or discomfort three days per month in the last three months associated with two or more of:
a) Improvement with defecation
b) Onset associated with a change in frequency of stool
c) Onset associated with a change in form of stool
The role of routine diagnostic investigation in patients with IBS:
Routine diagnostic investigation is based on the age of the patient, family history of selected organic diseases including colorectal cancer, inflammatory bowel disease(IBD), coeliac sprue and the presence of ‘alarm’ features(table1), such as rectal bleeding, weight loss, iron deficiency anaemia and nocturnal symptoms.(1) In patient with typical IBS symptoms and no alarm features, routine diagnostic investigation (complete blood count, serum chemistry, thyroid function tests, stool for ova and parasites and abdominal imaging) is not recommended(1)because of a low likelihood of uncovering organic disease.
Table-1 Lists of alarm features:
Rectal bleeding
Weight loss
Iron deficiency anaemia
Nocturnal symptoms: abdominal pain
family history of of selected organic diseases: colorectalcancer, Inflammatory Bowel Disease(IBD), celiac sprue
Summary of diagnostic investigation in patient with IBS : (1,2)
Diagnostic Investigations:
Routine serologic screening for coeliac sprue for patients with IBS-D and IBS-M.
Lactose Breath test done in lactose maldigestion despite dietary modification.
Colonoscopic Imaging done in IBS patient (>50 yrs age) with alarm feature to rule out organic diseases and screening of colorectal cancer.
Colonoscopy with random biopsies taken in IBS-D to rule out microscopic colitis.
Management of IBS:
The goal of IBS management is to provide relief of symptoms and improve overall well-being.(28)Most studies use a combination therapy including patient education and psychological therapies, diet and fibre therapy along with different types of new emerging pharmacological therapies.
Patient education and psychological therapies:
The majority of patients with IBS have anxiety, depression and features of somatization. Psychological therapies, including cognitive behavioral therapy, dynamic psychotherapy, hypnotherapy(1)shed new light on the management of patients with IBS. The outcome of psychological therapies is improved when delivered by a trained professional (physician, occupational therapist, nurse).(29) A study by Guthrie(30)showed that psychological therapy is feasible and effective in two thirds of patients with IBS who do not respond to standard medical treatment.
Role of diet in IBS:
The concept of food intolerance and the consequent elimination of certain foods from the diet benefit symptoms of IBS. However, there is no sufficient evidence to support this.(1)
Therapeutic effectof dietary fibre, bulking agents and laxatives:The quality of evidence supporting the recommended use of dietary fibre or bulking agents to regularize bowel function is poor.(31)Ispaghula husk(Psyllium hydrophilic mucilloid ) and calcium polycarbophil are moderately effective and can be given a conditional recommendation because of the weakest type of evidence.(1) Polyethylene glycol(PEG) laxative has a role in improving stool frequency but no effect on abdominal pain. Different clinical studies and expert opinion suggest that increased fibre intake may cause bloating, abdominal distension and flatulence.(32)So gradual adjustment of dose is advised for the use of these agents.
Therapeutic effectof antispasmodic agents including peppermint oil:
Certain antispasmodics (hyoscine, cimetropium,and pinaverium and peppermint oil) may provide short-term relief of abdominal pain/discomfort in IBS.(33,34)Evidence for safety and tolerability
Agent
Mechanism of action
Targeted disorder
Clinical status
Crofelemer
CFTR
IBS-D
Phase2b complete
Linaclotide
Guanylate cyclase-c agonist
IBS-C
Phase 3
Arverapamil
Calcium channel blocker
IBS-D
Phase 3
Asimadoline
Kappa opioid agonist
IBS
Phase 2b complete
Mitemcinal
Motilin receptor agonist
IBS-C
Phase 2
Ramosetron
5-HT 3 antagonist
IBS-D
Phase 3
TD-5108
5-HT 4 agonist
IBS-C
Phase 2
DDP-773
5-HT 3 agonist
IBS-C
Phase 2
DDP-225
5-HT 3 antagonist and NE reuptake inhibition
IBS-D
Phase 2
BMS-562086
Corticotropin-releasing hormone antagonist
IBS-D
Phase 2
GW876008
Corticotropin-releasing hormone antagonist
IBS
Phase 2
GTP-010
Glucagon-like peptide
IBS pain
Phase 2
AGN-203818
Alpha receptor agonist
IBS pain
Phase 2
Solabegron
Beta-3 receptor agonist
IBS
Phase 2
Espindolol (AGI-011)
Beta receptor antagonist
IBS (all subtypes)
Phase 2
Dextofisopam
2,3 benzodiazepinereceptors
IBS-D and IBS-M
Phase 3
Table 1: Source: ACG Task Force on IBS(2009)
of these agents are very limited.The commonest adverse effects are dry mouth,dizziness and blurred vision.(34-36)
Therapeutic effectof anti-diarrhoeal medications:
The anti-diarrhoeal agent ‘Loperamide’ is effective at slowing down colonic transit and improving stool consistency for the treatment of IBS-D with no severe adverse effects.(37)But safety and tolerability datas are still lacking in many studies.
Therapeutic effect of antibiotics:
Many studies show well tolerance of a short term course of non-absorbable antibiotics (Rifaximin) is most effective for improvement of global symptoms in IBS-D and IBS patient with the predominant symptom of bloating and other associated symptoms, such as diarrhoea and abdominal pain.(38-40) While, the Unted States Food and Drug Administration (FDA or USFDA) approved Rifaximin for treatment of traveler’s diarrhoea. Other antibiotics, Neomycin(41), Clarithromycin and Metronidazole(42)have been well evaluated for the management of IBS.
Therapeutic effect of Probiotics:
Probiotics have a large number of properties that can benefit IBS. Bifidobacteria is the active agent in probiotic combination therapy.Whereas many studies show Lactobacilli to have no impact on symptoms.(43)But one Korean study concluded that thecomposite probiotics containing Bifidobacterium bifidum BGN4, Lactobacillus acidophilus AD031, and other species are safe and effective, especially in patients who excrete normal or loose stools.(44) Recently, P Moayyedi and colleague in their systematic review recommend that probiotics appear to be efficacious in IBS patients ,but the magnitude of benefit and the most effective species and strain are uncertain.(45)
Therapeutic effect of the 5HT3 receptor antagonists:
Alosetron (5-HT3 receptor antagonists), with dosage of 0.5 to 1 mg daily, is more effective and the commonest drug used for treatment of patients with IBS-D in spite of serious side effects including constipation and colon ischemia.The balance model of benefits and harms for ‘Alosetron’ is most encouraging in women who have not responded to conventional therapies.(46,47)
Therapeutic effect of 5-HT4 receptor agonists:
Tegaserod (5-HT4 receptor agonist) is more effective for the treatment of IBS-C mostly in female and IBS-M. The side effects reported among the patient receiving Tegaserod are diarrhoea (commonest), cardiovascular events i.e. myocardial infarction, unstable angina, or stroke.(48,49)Currently Tegaserod is available from FDA through an emergency investigational new drug protocol. Other 5-HT4 agonists (Cisapride,Renzapride) have not demonstrated improvement compared with placebo.(50,51)
Therapeutic effect of the selective C-2 chloride channel activators:
Lubiprostone (selective C-2 chloride channel activator) is effective for relieving symptoms of IBS-C, mostly in women, and has less frequent side-effects including nausea(8%), diarrhea(6%) and abdominal pain(5%).(52)
Therapeutic effect of antidepressants :
Patients with prominent symptom of abdominal pain in IBS that fails to respond to peripherally acting agents often are considered for treatment with antidepressants (TCAs and SSRIs), however, limited data on safety and tolerability of these agents is shown.(53)Antidepressants have the combined effect of both central and peripheral mechanism in IBS.(54)SSRIs are better tolerated than TCAs and have a prokinetic effect hence work better in IBS-C.(53,55)whereas TCAs are of greater benefit for IBS-D.
Therapeutic effect of herbal therapies and acupuncture:
Unique Chinese herbal mixtures show a benefit in IBS management.(56) Traditional Chinese herbal remedies are routinely used in China to treat the condition, but so far have not been generally accepted by conventional Western medicine.(56,57)Bensoussanand colleague in one randomized, double-blind, placebo-controlledtrial concluded that the Chinese herbal formulations appear to offerimprovement in symptoms for some patients with IBS.(57) A systematic review of different trials of acupuncture was inconclusive because of heterogenous outcomes.(58,59) Hence further work is needed before any recommendations on acupuncture or herbal mixtures therapy.
Emerging therapies :
The improved understanding of underlying mechanisms in IBS is beneficial for the development of new pharmacological treatment options.
A brief overview of emerging agents in IBS therapy summarized in Table 1(1)
Conclusion:
IBS is a true medical disorder that has significant impact on those in agony with regard to symptom severity, disability, and impaired quality of life, which exceeds that of most GI disorders. Advances in research over the past several decades have paved the way for anameliorableunderstanding of the underlying pathophysiology and standardized symptom-based approaches that can be implemented in making a positive diagnosis and development of innovative treatment options for multiple IBS symptoms. Although many unanswered questions remain, the progress is promising and it has equipped physicians better to efficiently diagnose IBS and choose from a growing armamentarium of treatment options.
Syncope is a common condition encountered in acute medical practice. Many patients with syncope are initially labelled as having “collapse query cause”. It is defined as transient loss of consciousness (T-LOC) due to transient global cerebral hypoperfusion characterized by rapid onset, short duration, and spontaneous complete recovery1. Incidence of syncope is difficult to determine accurately as many cases remain unreported. Some studies quote an overall incidence rate of a first report of syncope to be 6.2 per 100 person-years. Clearly this is age related and the incidence increases dramatically in patients over the age of 70 years2. Syncope accounts for 1-6% of hospital admissions and 1% of emergency department (ED) visits per year3-5. Hospital episode statistics from NHS hospitals in England reported a total of 119,781 episodes of collapse/syncope for the financial year 2008-09 which is about twice the number of episodes reported in the year 1999-2000. About 80% of patients were admitted and they have an average length of stay of 3 days accounting for over 269,245 bed days during that financial year6.
Syncope is also associated with significant mortality and morbidity if left untreated. Literature reports a 6-month mortality of 10%, which can go up to 30% if cardiac syncope is untreated7. Non-cardiac syncope is associated with a survival rate comparable to people with no syncope2. Syncope is also a risk factor for fractures related to falls especially in elderly and can cause significant morbidity in this group8. In addition, there are significant health care related costs associated with management of syncope. Cost per diagnosis can vary from over £611 in the UK to €1700 in Italy. Hospitalisation alone accounted for 75% of cost in some studies9,10. Diagnosis of this condition can be difficult especially if there is a lack of structured approach. Over the last few years this topic has attracted enormous interest and several studies have been published, aiming at improving the approach to this condition. Standardised syncope pathways improve diagnostic yield and reduced hospital admissions, resource consumption and over all costs10. Recently the task force for the diagnosis and management of syncope of the European Society of Cardiology published guidelines for the diagnosis and management of syncope1. However, in spite of the available evidence very few hospitals have standardised syncope pathways for the management of this complex condition. Only 18% of EDs have specific guidelines and access to a specialist syncope clinic11. This article focuses on evidence based structured evaluation of syncope. Current practice in the management of syncope Due to the difficulty in diagnosis and mortality associated with this condition, a cautious approach may be taken by physicians resulting in hospitalisation of majority of patients presenting with syncope. We recently audited the practice of syncope in our hospital, which is a tertiary centre in the north of Scotland. 58 patients admitted with this condition over a period of a month were included in the audit. It showed an average length of stay (LOS) of 4.76 days in these patients. Due to a lack of methodical approach and standardised pathway for management of this condition many patients were subjected to several inappropriate inpatient investigations significantly prolonging the LOS and increasing the cost. Only 7 (12%) cardiac events were observed in this group and in retrospect a good methodical approach would have predicted these events. It should be noted that even in the geriatric population, reflex syncope that carries a benign prognosis is more common than cardiac syncope2. A systematic approach to the management of syncope (Figures 1 and 2). The causes of syncope can be broadly divided in to cardiac causes and non-cardiac causes (Table 1). Initial evaluation leads to a diagnosis in less than 50% patients in most instances4,12-14. If there is uncertainty about diagnosis then the patient is risk stratified. High-risk patients are hospitalised, evaluated and treated whereas early discharge could be considered in low risk patients. Aetiology of Syncope41
Neurally-mediated (Reflex) Syncope
Cerebro vascular
Vasovagal syncope
Carotid sinus syncope
Situational syncope
e.g., Micturition, post prandial, defecation, cough
Relevant blood tests (e.g. to rule out metabolic abnormality)
Pacemaker check if appropriate
History Many patients with syncope are initially labelled as having “collapse query cause”. Loss of postural tone is termed “collapse”. Indeed, the term “collapse query cause” does not give any useful information regarding the underlying condition. A clear history from the patient and the bystander or witness (if available) is the key to the diagnosis. Firstly, determine if the collapse was associated with loss of consciousness (LOC). LOC can be transient (T-LOC) or prolonged. Categorising “collapse” is important at this stage as the aetiology and approach to each category is different (Figure 1). Secondly, establish if the collapse was syncopal. The LOC should be transient (e.g. did the patient regain consciousness in the ambulance, before or on arrival to hospital?), of rapid onset and associated with a spontaneous complete recovery. Also the mechanism should be due to transient global hypoperfusion. T-LOC secondary to other mechanisms such as trauma and brief seizures should be excluded. On occasions syncope could be associated with brief jerking movements mimicking seizures15. Also note that a transient ischemic attack (TIA), commonly listed as a differential diagnosis of syncope by physicians, is not a cause of syncope as this is not associated with global cerebral hypoperfusion. The absence of a coherent history because patient had no recollection of events and there was no witness account available can make this distinction difficult. This is also particularly difficult in the elderly with cognitive impairment. Other useful information includes whether the syncope was associated with postural change. Orthostatic hypotension occurs after standing. If present it will be useful to check drug history (new vasodepressive drugs). Features suggestive of Parkinson’s disease or amyloidosis may raise the possibility of autonomic neuropathy. A strong family history of sudden cardiac death may be of relevance. Table 3 summarises the features of neurally mediated and cardiac syncope. Table 3 Features suggesting neurally mediated and cardiac syncope42
Neurally mediated
Cardiac
Preceded by prodrome
Related to particular activity - e.g., Micturition, postprandial, prolonged standing, unpleasant situations
Associated with nausea and vomiting
After exertion
Absence of prodrome, no warning
Associated with chest pain, breathlessness, palpitation
During exertion or supine
History of cardiac disease
Family history of sudden cardiac death
Physical examination The next step is a thorough physical examination. This should include an ABC approach if the patient is very ill and particular attention should be given to exclude immediate life threatening conditions such as pulmonary embolism, acute myocardial infarction, life threatening arrhythmias, acute aortic dissection, seizures etc… Recording the vital signs is important as it may give a clue to diagnosis (e.g., acute hypoxia may indicate massive pulmonary embolism). Recording postural blood pressure when lying and during active standing for 3 minutes is useful to exclude orthostatic hypotension1. Recording a deficit in blood pressure in both arms may be a useful clinical finding especially if acute aortic dissection is suspected. Thorough cardio respiratory examination may reveal an obvious condition such as cardiac failure or aortic stenosis. Patients should also be examined for potential injuries as a result of syncope. Standard ECG A 12 lead ECG should be performed in all patients admitted with syncope. The abnormalities in table 4 would suggest a cardiac aetiology. The QT interval should always be measured, as it is a commonly overlooked abnormality. Blood tests Blood tests are usually unhelpful in establishing a diagnosis but can detect metabolic abnormalities such as hypoglycaemia, electrolyte abnormalities and other causes to explain LOC especially when witness account is not available. An acute drop in haemoglobin suggests blood loss. One recent study claims the usefulness of brain natriuretic peptide (BNP) for predicting adverse outcomes in syncope but it is not externally validated yet and it is too early to recommend for routine clinical practice16. Pacemaker check It is not uncommon to see a patient with a pacemaker implanted, admitted to hospital with syncope. In these circumstances, it is essential to rule out a device malfunction although this is not a common cause of syncope. A preliminary and easy test will be interrogating the pacemaker if available. This should pick up any problems with the pacemaker in most instances. With the above information establishing a diagnosis will be possible in a significant proportion of patients. Further investigations and management should be guided by the underlying diagnosis1. However in over half of patients the diagnosis may still be uncertain12,13,17. The following section explains the management of unexplained syncope. Risk stratification in patients with unexplained syncope (Tables 4 and 5)Table 4 ECG changes in ‘high-risk’ Syncope41
ECG changes favouring bradyarrhythmias
High degree AV blocks – Mobitz type 2 second degree AV block, complete heart block, trifascicular block (first degree heart block with left bundle branch block (LBBB) or right bundle branch block (RBBB) with axis deviation)
Bifascicular block (defined as either LBBB or RBBB combined with left anterior fascicular block or left posterior fascicular block) especially if new
Other intraventricular conduction abnormalities (QRS duration >0.12 s)
Asymptomatic sinus bradycardia (<50 bpm), sinoatrial block or sinus pause >3 s in the absence of negatively chronotropic medications
ECG changes favouring tachyarrhythmias
Pre-excited QRS complexes (e.g. WPW syndrome)
Prolonged QT interval
Right bundle branch block pattern with ST-elevation in leads V1–V3(Brugada syndrome)
Negative T waves in right precordial leads, epsilon waves and ventricular late potentials suggestive of arrhythmogenic RVD
Q waves suggesting myocardial infarction
Non sustained Ventricular Tachycardias
Table 5 – Clinical features of high-risk syncope1,18-23
History of severe structural heart disease or heart failure, presence of ventricular arrhythmia
Syncope during exertion or supine
Absence of prodrome or predisposing or precipitating factors
Preceded by palpitation or accompanied by chest pain or shortness of breath
Family history of sudden cardiac death
Examination suggestive of obstructive valvular heart disease
Syncope associated with trauma
Systolic blood pressure less than 90mm Hg
Hematocrit less than 30% (acute drop in hemoglobin)
When the cause of syncope is uncertain it is essential to risk stratify patients to enable appropriate treatment and further investigation. Risk stratification tools There are several scoring systems for risk stratification of syncope. Syncope Evaluation in the Emergency Department Study (SEEDS), Osservatorio Epidemiologico sulla Sincope nel Lazio (OESIL score), Evaluation of Guidelines in SYncope Study (EGSYS score), San Francisco Syncope Rule (SFSR), The Risk stratification Of Syncope in the Emergency department (ROSE) and American College of Emergency Physicians clinical policy are the popular ones and each has its own advantages and disadvantages1,16,18-23. Discussing each scoring system is beyond the scope of this article and we shall restrict the discussion to the summary of these risk stratification tools (Table 5). It will be too early to include all the factors mentioned in the ROSE study, as it is not externally validated yet. It could be argued that taking all the risk factors described may increase admission rates but this approach may at least not miss the high-risk patient. This is a developing field and more evidence is likely to be published soon. High-risk vs. low-risk syncope: A high-risk syncope patient is the one where a cardiac cause is likely and where the short-term mortality is high due to major cardiovascular events and sudden cardiac death. High-risk syncope is said to be present if any of the features in the table 4 or 5 are present. Management of low-risk syncope Patients with a single or very infrequent syncope are usually reassured and discharged, as the short-term mortality is low1,2. Tilt table test is not usually required where a single or rare episode of neurally mediated syncope is diagnosed clinically. One exceptional circumstance where single rare episodes are investigated further with a tilt table test is when there could be an occupational implication (e.g. aircraft pilot) or if there is a potential risk of physical injury. Patients with recurrent unexplained syncope need to be further investigated (see below). Management of high-risk syncope / suspected cardiac syncope High-risk patients usually require hospitalisation and inpatient evaluation. Other high-risk patients who may be considered for admission are vulnerable patients susceptible to serious injuries, for example, elderly patient or a patient with multiple co-morbidities. Further investigations (Table 6)
Non invasive
Invasive
Echocardiography
ECG monitoring
Telemetry
Holter monitoring
External loop recorder*
Carotid sinus massage
Cognitive testing (in elderly)
Ambulatory blood pressure monitoring
Tilt table test*
Exercise stress test
Implantable loop recorder*
Coronary angiography*
Electrophysiology*
* Specialist Investigation Echocardiography Echocardiography is a relatively inexpensive and non-invasive investigation. It should be performed if there is a clinical suspicion of a significant structural abnormality of heart such as ventricular dysfunction, outflow tract obstruction, obstructive cardiac tumours or thrombus, pericardial effusion etc… The yield of this test is low in the absence of clinical suspicion of structural heart disease. However in the presence of a positive cardiac history or an abnormal ECG, one study detected LV dysfunction in 27% of patients and half of these patients had syncope secondary to an arrhythmia. In patients with suspected obstructive valvular disease 40% had significant aortic stenosis as a cause of syncope24. ECG monitoring These tests have utility in identifying arrhythmogenic syncope. If a patient has syncope correlating with a significant rhythm abnormality during the monitoring period with the device, then the cause of syncope is due to the underlying rhythm abnormality. On the other hand, if no rhythm abnormality is recorded during a syncopal attack, then an underlying rhythm problem as a cause of syncope is excluded. Therefore, these tests are meaningful only if there is a symptom-rhythm correlation, which is the working principle of these devices. In the absence of syncope, during the monitoring period, these tests may pick up other abnormalities that may be relevant. For example, rapid prolonged supra-ventricular tachycardias, ventricular tachycardias, periods of high degree AV blocks (mobitz type 2 or complete heart block) or significant sinus pauses >3seconds (except during sleep, negatively chronotropic therapy and trained athletes), which will require further investigation or treatment. Telemetry Telemetry can be used in inpatients. Although the diagnostic yield of this investigation is only 16%, given the high short-term mortality, this test is indicated in the high-risk group 1. Usually patients are monitored for 24 to 48 hours although there is no agreed standard period for monitoring25. Holter monitoring This involves connecting the patient through cutaneous patch electrodes. It records the ECG activity conventionally over 24-48 hours or at times up to 7 days. It is particularly useful only in patients who have frequent regular symptoms (≥1 per week). For this reason, the yield of this test can be as low as 1-2% in unselected population1. Long inpatient waiting lists in some hospitals can significantly prolong the length of stay and cost. Selecting patients carefully for this test based on risk stratification will reduce costs and waiting lists. Carotid sinus massage This simple bedside test is indicated in patients over the age of 40 years with syncope of unexplained origin after initial evaluation. A ventricular pause lasting >3 s and/or a fall in systolic BP of >50mmHg defines carotid sinus hypersensitivity (CSH) syndrome. It is contraindicated in patients with recent cerebrovascular accidents (past 3 months) or with carotid bruit except when a Doppler study has excluded significant stenosis1. Cognition test If an elderly patient had forgotten about the events, in the absence of an obvious cause, it may be useful to test cognition. If cognitive impairment is present, common problems associated with cognitive dysfunction should be considered e.g. falls, orthostatic hypotension. Other investigations In spite of the above tests if a cause is not determined, early specialist input is recommended for further investigation and treatment. The following non-invasive and invasive investigations may be appropriate in these circumstances. An external loop recorder This is a non-invasive form of electrocardiographic monitoring. The principle is same as that of Holter monitoring. External loop recorders have a loop memory that continuously records and deletes ECG. When activated by the patient, typically after a symptom has occurred, 5 – 15 min of pre-activation ECG is stored and can be retrieved for analysis. Studies have shown that they have increased diagnostic yield compared to Holter1. They should be considered in patients who have symptoms on a monthly basis. A Tilt table test This is indicated in cases of recurrent unexplained syncope after relevant cardiac causes of syncope are excluded and a negative Carotid sinus massage performed in the absence of contraindications. It is also indicated when it is of clinical value to demonstrate patients susceptibility to reflex syncope and thereby to initiate treatment. Other less common indications are recurrent unexplained falls, differentiate jerking movements secondary to syncope and epilepsy, diagnose psychogenic pseudo syncope and differentiate orthostatic and reflex syncope. Indication of this test in the context of a single unexplained syncope is discussed above. Ambulatory blood pressure monitoring This may be useful in patients with unexplained syncope particularly in old age to check if there is an element of autonomic failure and if a single set of orthostatic blood pressure recording is not helpful. In one study, it has been shown that 25% of the elderly patients admitted with falls or syncope had postprandial hypotension especially after breakfast26. It may be more readily available than a tilt table test in some centres. Exercise stress test This may be useful in a rare entity called exercise induced syncope. Outflow tract obstruction should be excluded by echocardiography before subjecting a patient to this test especially in the presence of relevant signs. However there is no evidence for supporting this test in investigating syncope in general population. Implantable loop recorders These are implanted subcutaneously. It needs to be activated either by the patient or a bystander after a syncopal attack. It is indicated in high-risk patients where a comprehensive evaluation did not establish an underlying diagnosis. In the absence of high risk factors, it is also indicated in patients with recurrent unexplained syncope especially if infrequent. Conventionally it is used as a last resort in patients with recurrent unexplained syncope as the initial costs are high. It has been shown in one study to be more cost effective than the conventional strategy and was more likely to provide a diagnosis in patients with recurrent unexplained syncope27. However patients with poor LV function and those at high risk of life-threatening arrhythmias were excluded from this study. Coronary angiography or CT coronary angiography This may be helpful in suspected myocardial ischemia or ischemia related arrhythmias. Electrophysiological study may be considered in certain circumstances by cardiologists. When a standardised pathway is used, diagnosis is ascertained in 21% patients on initial evaluation and further 61% patients with early investigations. Only in 18% patients the diagnosis was still uncertain12. Other studies have shown similar results28. Although these results are from a dedicated syncope unit following a standardised pathway, these could be extrapolated to any unit following these standardised pathways. Further management is dictated by the underlying diagnosis with early specialist input for appropriate treatment. Treatments Single or rare episodes of reflex syncope do not require treatment. However, recurrent troublesome reflex syncope may warrant treatment. Treatment modalities are primarily non-pharmacological such as tilt training, physical counter pressure manoeuvres (leg crossing, hand gripping) and ensuring adequate hydration29. If refractory to non-pharmacologic measures midodrine (alpha agonist) may be considered in patients with frequent hypotensive symptoms30,31. Fludrocortisone may be used in elderly but there is no trial evidence to support this. Betablockers have been presumed to lessen symptoms but are shown to be ineffective in several studies 32. They may potentially exacerbate bradycardia in carotid sinus syncope and are not recommended in treatment of reflex syncope. Treatment with cardiac pacing in reflex syncope is controversial and may be considered in patients with predominant cardio inhibitory response on carotid sinus massage (in CSH syndrome) or on tilt test (in reflex syncope). It should be noted that cardiac pacing has no effect on the often-dominant vasodepressor component of reflex syncope. In patients with orthostatic hypotension, non-pharmacologic measures like increased salt and water intake, head up tilt sleeping, physical counter pressure manoeuvres, abdominal binders and compression stockings may help reducing symptoms. Midodrine is an efficient alternative in these circumstances and fludrocortisone also can be used.33,34Syncope secondary to cardiac arrhythmias needs treatment if a causal relationship is established. Potential reversible causes such as electrolyte abnormalities and drug induced causes should be excluded. Cardiac pacing is a modality of treatment in significant bradyarrhythmias secondary to sinus node or advanced AV nodal disease such as mobitz type 2 block, complete heart block or tri-fascicular block. Catheter ablation and anti-arrhythmic drug therapy are the main modalities of treatment for tachyarrhythmias. Implantable cardioverter defibrillator may be indicated in patients susceptible to malignant ventricular tachyarrhythmias. Treatment of syncope secondary to structural cardio pulmonary abnormality will need surgical intervention if possible. Driving and Syncope Doctors are poor at addressing and documenting this issue35. Table 7 gives some useful information from the DVLA website (http://www.dft.gov.uk/dvla/medical/ataglance)36. This information is country specific and subject to change. Table 7 – Driving and Syncope in the UK36
Type of Syncope
Group 1 entitlement (car, motorcycle etc.,)
Group 2 entitlement (Large goods vehicle, passenger carrying vehicle)
Simple faint
No restrictions
No restrictions
Unexplained syncope with low risk of recurrence*
Allowed to drive 1 month after the event
Allowed to drive 3 months after the event
Unexplained syncope with high risk of recurrence** and cause identified and treated
Allowed to drive 1 month after the event
Allowed to drive 3 months after the event
Unexplained syncope with high risk of recurrence** and cause not identified
Licence is refused or revoked for 6 months
Licence is refused or revoked for 12 months
*Absent clinical evidence of structural heart disease and normal ECG** Abnormal ECG, clinical evidence of structural heart disease, syncope causing injury, recurrent syncope Syncope unitsSyncope units aim to evaluate syncope (and related conditions) in dedicated units consisting of generalists and specialists with an interest in syncope. A sufficient number of patients are required to justify such a unit. They are well equipped with facilities for recording ECG, blood pressures, tilt table, autonomic function testing, ambulatory blood pressure monitoring, and invasive and non-invasive electrocardiographic monitoring. It has been shown to be cost effective and reduces health care delivery costs by reducing admission rates, readmission rates and event rates. Examples include the Newcastle model, Manchester model and the Italian model.12,18,37,38Conclusions The incidence of syncope is increasing in the UK with an aging population. There is significant cost incurred in the delivery of health care for this condition. The approach to syncope varies widely amongst practising physicians due to lack of a methodical approach. A thorough initial evaluation yields a diagnosis in less than half of the patients. When the cause of syncope remains unexplained after initial evaluation, the patients should be risk stratified. While a patient with a single episode of low risk syncope can be reassured and discharged, those with high-risk features should be hospitalised for further management. Outpatient evaluation could be offered for low risk patients if recurrent. Early specialist input should be sought in high-risk syncope and recurrent unexplained syncope. This standardised approach or pathway will reduce cost by reducing hospitalisation, inappropriate investigations and length of stay.
Key Facts
Collapse associated with transient loss of consciousness is called syncope if it is due to transient global cerebral hypoperfusion and characterized by rapid onset, short duration, and spontaneous complete recovery
Standardised syncope pathways improve diagnostic yield and reduce hospital admissions, resource consumption and over all costs
A thorough initial evaluation yields a diagnosis in less than half of patients. If the cause of syncope is undetermined after initial evaluation, patients should be risk stratified
Early discharge should be considered in low risk patients while high-risk patients need urgent evaluation.
Early specialist referral is recommended in patients with high risk syncope and recurrent unexplained syncope
Future Interests Syncope had been known for several decades and still remains a complex condition, as the exact mechanisms are poorly understood especially in non-cardiac syncope. Mechanism of syncope in the elderly patients may be different from those of young patients and studies should focus in understanding the mechanics. Further research is needed in risk stratifying syncope. It may enable us to develop more robust care pathways for management of syncope. The role of BNP in investigating and risk stratifying syncope need to be further clarified. In spite of sophisticated tests the cause of syncope in a proportion of patients remain uncertain. Studies should focus on the long-term outcome and management of syncope in this group. The role of implantable loop recorder in the investigation of syncope should be better defined and more studies should focus on when it should be offered in the pathway of management of syncope. Studies are also required to develop effective pharmacotherapies for this condition.
Introduction: Most experts define infertility as not being able to get pregnant after at least one year of trying. Women who are able to get pregnant but then have recurrent miscarriages are also said to be infertile. The infertility definition made a difference. The World Health Organization definition based on 24 months of trying to get pregnant is recommended as the definition that is useful in clinical practice and research among different disciplines.1Magnitude of the Problem: It is a growing problem and across virtually all cultures and societies almost all over the World and affects an estimated 10%-15% of couples of reproductive age. In recent years, the number of couples seeking treatment for infertility has dramatically increased due to factors such as postponement of childbearing in women, development of newer and more successful techniques for infertility treatment, and increasing awareness of available services. This increasing participation in fertility treatment has raised awareness and inspired investigation into the psychological ramifications of infertility. Consideration has been given to the association between psychiatric illness and infertility. Researchers have also looked into the psychological impact of infertility per se and of the prolonged exposure to intrusive infertility treatments on mood and well-being. There is less information about effective psychiatric treatments for this population; however, there is some data to support the use of psychotherapeutic interventions2. Why infertility has a psychological effect on the couple? Parenthood is one of the major transitions in adult life for both men and women. The stress of the non-fulfilment of a wish for a child has been associated with emotional squeal such as anger, depression, anxiety, marital problems and feelings of worthlessness. Partners may become more anxious to conceive, ironically increasing sexual dysfunction and social isolation. Marital discord often develops in infertile couples, especially when they are under pressure to make medical decisions. Couples experience stigma, sense of loss, and diminished self-esteem in the setting of their infertility3. Male and female partner respond differently: In general, in infertile couples women show higher levels of distress than their male partners4; however, men’s responses to infertility closely approximate the intensity of women’s responses when infertility is attributed to a male factor3. Both men and women experience a sense of loss of identity and have pronounced feelings of defectiveness and incompetence. Women trying to conceive often have clinical depression rates similar to women who have heart disease or cancer. Even couples undertaking IVF face considerable stress. Emotional stress and marital difficulties are greater in couples where the infertility lies with the man. Therefore the psychological impact of infertility can be devastating to the infertile person and to their partner. Factors influencing psychological stress: According to one study done in Sweden, three separate factors seem to contribute to the psychological stress men and women experience as a result of their infertility. The three factors, in order of importance for the women were,1. "Having Children is a Major Focus of Life"2. "The Female Role and Social Pressure"3. "Effect on Sexual Life"The men in the study reversed the order of importance of factors 1 and 2. The third factor was equally significant to both the men and women. It was also shown that women experienced their infertility more strongly than the men. Women also showed a more intense desire to have a baby than men.5.Behaviour of the couple as a result of infertility: Stress, depression and anxiety are described as common consequences of infertility. A number of studies have found that the incidence of depression in infertile couples presenting for infertility treatment is significantly higher than in fertile controls, with prevalence estimates of major depression in the range of 15%-54%6,7,8,9. Anxiety has also been shown to be significantly higher in infertile couples when compared to the general population, with 8%-28% of infertile couples reporting clinically significant anxiety9,10. The causal role of psychological disturbances in the development of infertility is still a matter of debate. A study of 58 women from Lapane and colleagues reported a 2-fold increase in risk of infertility among women with a history of depressive symptoms; however, they were unable to control for other factors that may also influence fertility, including cigarette smoking, alcohol use, decreased libido and body mass index11. Psychological factors may also affect the reproductive capacity: Although infertility has an effect on a couple’s mental health, different psychological factors have been shown to affect the reproductive ability of both partners. Proposed mechanisms through which depression could directly affect infertility involve the physiology of the depressed state such as elevated prolactin levels, disruption of the hypothalamic-pituitary-adrenal axis, and thyroid dysfunction. One study of 10 depressed and 13 normal women suggests that depression is associated with abnormal regulation of luteinizing hormone, a hormone that regulates ovulation12. Changes in immune function associated with stress and depression may also adversely affect reproductive function13. Further studies are needed to distinguish the direct effects of depression or anxiety from associated behaviours (e.g., low libido, smoking, alcohol use) that may interfere with reproductive success. Since stress is also associated with similar physiological changes, this raises the possibility that a history of high levels of cumulative stress associated with recurrent depression or anxiety may also be a causative factor. Result of treatment: While many couples presenting for infertility treatment have high levels of psychological distress associated with infertility, the process of assisted reproduction itself is also associated with increased levels of anxiety, depression and stress14. A growing number of research studies have examined the impact of infertility treatment at different stages, with most focusing on the impact of failed IVF trials15. Comparisons between women undergoing repeated IVF cycles and first-time participants have also suggested that ongoing treatment may lead to an increase in depressive symptoms16. The data, however, is still controversial since other studies have found minimal psychological disturbance induced by the infertility treatment process or IVF failure17,18. In light of the discrepancy in results, there has been increasing interest in the factors that contribute to drop out from infertility treatment since this population is often not included or decline to participate in studies. Whereas cost or refusal of physicians to continue treatment have been cited as reasons for discontinuing treatment, recent research suggests that a significant number of drop outs are due to psychological factors19,20,21. The outcome of infertility treatment may also be influenced by psychological factors. A number of studies have examined stress and mood state as predictors of outcome in assisted reproduction. The majority of these studies support the theory that distress is associated with lower pregnancy rates among women pursuing infertility treatment7,16,22,23,24,25. Conclusion: In light of all the data suggesting that psychological symptoms may interfere with fertility, success of infertility treatment and the ability to tolerate ongoing treatment; interest in addressing these issues during infertility treatment has grown. Since psychological factors play an important role in the pathogenesis of infertility, exploration of this is also an important task to manage this devastating problem, which has cultural and social impact.
Vitiligo is one of the oldest and commonest skin disorders affecting approximately 1-2% of the human population.1 The disease shows no regard to the ethnic, racial or socioeconomic background of the affected sufferers. The cosmetic impact of this disease is tremendous and its psychological impact devastating particularly in coloured races.2,3,4 The aetiopathogenesis of this disease is now much better understood (table 1)5 compared with a decade earlier but much remains unknown. In parallel with these developments on the aetiological front, a lot of new advances have been made on the therapeutic front as well. With these new therapeutic options, we are currently in a much better position to treat this disease than we were a decade or two earlier. So, how far and how satisfactorily are we able to treat this disorder now? What are the new treatment options available for this disorder and how far have they helped a dermatologist to claim a cure for this disorder? These are some of the questions that will be addressed in this paper.
New advances in management
Medical therapies
The most recent advances on the medical front have been Narrowband Ultraviolet B (NB-UVB) therapy, Targeted Ultraviolet B (UVB), Excimer laser therapies, topical immunomodulator treatment in the form of topical calcineurin inhibitors, topical pseudocatalase, and topical Vitamin D analogues in combination with Ultraviolet (UV) light.
NB-UVB
NB-UVB, using UV-lamps with a peak emission of around 311nm has now emerged as the treatment of first choice in generalized vitiligo as well as vitiligo vulgaris (patchy vitiligo).6,7,8 The efficacy of NB-UVB in vitiligo was first demonstrated by Westerhof and Nieuwboer-Krobotova in 1997.9 Since then there have been a large number of clinical studies that have demonstrated the therapeutic benefit of NB-UVB in vitiligo patients. The mechanism of action of NB-UVB in vitiligo is through induction of local immunosuppression and stimulation of the proliferation of melanocytes in the skin and the outer root sheath of hair follicles.6 There is a stimulatory effect on melanogenesis and on the production of Melanocyte Stimulating Hormone (MSH).6 Comparison studies have shown a significantly enhanced rate of repigmentation with NB-UVB compared with topical Psoralen and Ultraviolet A (PUVA) therapy.10 Furthermore, the incidence of adverse effects seen commonly with topical PUVA, such as phototoxicity, is significantly reduced with the use of NB-UVB.
NB-UVB has shown a number of advantages over PUVA in vitiligo patients in addition to its excellent efficacy. These advantages include its extremely low side-effect profile particularly on the systemic front, its established safety in children, and safety in pregnant females. NB-UVB also has considerably better patient compliance as there is no need to time the exposure with any drug intake or any need for eye protection beyond treatment exposure time. A recent double-blind randomized11 study comparing NB-UVB with PUVA demonstrated a much better efficacy with NB-UVB. The study found that repigmentation achieved with NB-UVB was much better with respect to colour matching with uninvolved skin, and this was also more persistent than that achieved with PUVA.11
In addition NB-UVB has been used in childhood vitiligo with excellent results.12 No additional adverse effects were seen in children with NB-UVB as compared with those in adults. Furthermore, given the long-term safety profile of NB-UVB in comparison with PUVA as far as skin malignancies are concerned,13 NB-UVB is now preferred over all other treatment options in the management of generalized vitiligo in both adults and children.
Table 1: Aetiological hypothesis of vitiligo5
Aetiological hypothesis
Brief explanation
Autoimmune hypothesis
Believes that vitiligo occurs because of destruction of melanocytes by an immune mechanism.
Most favoured theory at present, supported by many recent in-vitro studies.
Auto-cytotoxic hypothesis
Believes that vitiligo occurs because of accumulation of toxic metabolites in the melanocytes secondary to a defect in their metabolic clearance of the toxins.
Neurogenic hypothesis
Believes that vitiligo is because of an altered reaction to neuropeptides, catecholamines and their metabolites by epidermal melanocytes.
Biochemical hypothesis
Believes that over-secretion of hydrobiopterin, a cofactor of tyrosine hydroxylase results in accumulation of catecholamines that in turn results in formation of reactive oxygen species in the melanocytes. These reactive oxygen species are thought to cause destruction of affected melanocytes in vitiligo patients.
NB-UVB has been used in combination with different topical agents to increase its efficacy and thus shorten the total duration of treatment. Treatment options that have been used with NB-UVB in vitiligo till date include topical tacrolimus,14,15 pimecrolimus,16 Vitamin D analogues17,18 and even topical pseudocatalase.19 While some studies have shown a synergistic effect with these combinations, others have found the efficacy of the combinations to be similar to NB-UVB alone. In one half-body comparison study, topical placental extract was used in combination with NB-UVB but the combination was shown to offer no added benefit than NB-UVB alone.20 Therefore, the ideal topical agent to be combined with NB-UVB remains unknown.
Laser Therapy
Excimer laser, which uses Xenon-Chlorine (Xe-Cl) gas and produces a monochromatic laser light of 308nm wavelength, is another innovative treatment option for vitiligo. The laser system has been used with increasing frequency over the last few years for targeted treatment of individual vitiligo lesions.21 The laser is used either alone or in combination with topical immunomodulator or PUVA-sol therapy.22,23 Treatment with this laser is claimed to give extremely good and early results in both localized and segmental vitiligo. In a pilot study21 on 18 patients with 29 affected areas 57% of lesions showed varying degrees of repigmentation after just six exposures over two weeks. The figure was increased to 87% after 12 treatments over four weeks.21 Another recent study has reported a repigmentation of >75% in 61% of lesions after 30 treatments with Excimer laser. Repigmentation was found to be better on the face and trunk than on the extremities.24
Topical therapies, particularly topical tacrolimus, have been used in combination with Excimer laser. This combination has been claimed to be more effective than Excimer laser alone.22 In a randomized right-left comparison study22 with 14 patients, Excimer light monotherapy was compared with a combination of Excimer laser with topical tacrolimus. While 20% of lesions treated with Excimer laser alone achieved >75% repigmentation, the same degree of repigmentation was obtained in 70% lesions with the combination treatment.22 Topical methoxsalen has also been used in combination with Excimer laser phototherapy and this has been claimed to have worked better than laser therapy alone.23
The advantage of Excimer laser therapy over conventional UVB therapy is the targeted mode of treatment with no exposure of the uninvolved skin. Moreover, the onset of repigmentation is earlier with Excimer laser therapy than with UVB therapy.
Targeted UVB therapy
This is another recent innovation in vitiligo management that has arrived over the last few years. The beauty with this therapy is that it delivers high intensity UVB light only to the affected vitiliginous areas, avoiding any exposure to the uninvolved skin. This not only decreases the cumulative UVB dose received by an individual patient, but is also claimed to improve the efficacy of treatment quite significantly.
Targeted UVB therapy, as expected, finds its use more in the treatment of focal and segmental types of vitiligo. In fact, the first study25 with targeted UVB therapy was done on eight patients with segmental vitiligo. Five of these patients achieved >75% repigmentation of their lesions with this therapy.25
Targeted UVB therapy offers certain advantages over Excimer laser phototherapy. The treatment is safer and more efficacious compared with conventional UVB therapy, and almost as efficacious but much less costly than Excimer laser therapy.26
Systemic immunomodulator therapy
Vitiligo is thought to be an immune-mediated disease and thus immune-suppressive and immunomodulator agents have been used on a regular basis in this disease. Among the immunosuppressants, systemic steroids have been the most commonly used. However, systemic steroid therapy has always been associated with a high incidence of adverse effects especially in children which is the age-group most commonly affected. To overcome this limitation, steroids have been given in pulse or even in mini-pulse form. A prospective study involving 14 patients with progressive or static vitiligo showed cessation of disease activity and a repigmentation rate of 10-
60% after high-dose methylprednisolone pulse therapy administered on three consecutive days.27 Systemic steroids have also been administered in a mini-pulse form on two consecutive days every week, known as Oral Minipulse (OMP) therapy. The first study demonstrating the efficacy of OMP with oral betamethasone (0.1mg/kg with a maximum of 5mg) was described in 1991.28 In a later study29 on childhood vitiligo, betamethasone was replaced by oral methylprednisolone and combined with topical fluticasone ointment on the vitiligo lesions. The disease was arrested in >90% of patients, and >65% of children achieved good to excellent (>50%) repigmentation of their vitiligo lesions.29
Topical Vitamin D analogues
Vitamin D analogues, particularly Calcipotriol, have been used topically either alone or in combination with topical steroids in the management of vitiligo. The basis for the use of these agents is that Vitamin D3 affects the growth and differentiation of both melanocytes and keratinocytes. This has been further proved by the demonstration of receptors for 1 alpha-dihydroxyvitamin D3 on the melanocytes. These receptors are believed to have a role in stimulating melanogenesis.29 Vitamin D analogues have given variable results in the treatment of vitiligo in different studies. These agents have also been used in combination with UV-light (including NB-UVB) and topical steroids with variable results.30,31,32
Topical immunomodulators
Topical immunomodulators, such as tacrolimus and pimecrolimus, have been the most promising recent additions to topical vitiligo therapy. In fact because of their efficacy and a remarkable safety profile the use of these agents in vitiligo has shown a consistently increasing trend over the last few years. These agents can be safely administered in young children, as they don’t cause any atrophy or telangiectasia of the skin even after prolonged use. There is also no risk of hypothalamic-pituitary-adrenal (HPA) axis suppression as seen with the widespread use of potent topical steroids.33 The first study that demonstrated the efficacy of tacrolimus in vitiligo was published in 2002.34 In this study tacrolimus was used in six patients with generalized vitiligo and five of them achieved >50% repigmentation of their lesions by the end of study period.34 Since then many additional studies have been published on this subject and have clearly demonstrated the role of topical tacrolimus in vitiligo. The best results with topical immunomodulator therapy have been seen on exposed parts of the body such as the face and neck and, as with any other therapy, the acral parts of the body respond the least.34,35 Similar results were obtained with the use of topical pimecrolimus in vitiligo patients.36
Pseudocatalase
Pseudocatalase has been used in combination with Dead Sea climatotherapy or UVB exposure for the treatment of vitiligo. The basis for the use of this agent in vitiligo is the evidence of oxidative stress and high H2O2 levels in the lesional skin.37 While some earlier studies37 demonstrated excellent results with this agent in inducing repigmentation in vitiligo, later studies have cast doubts on its efficacy.38 Pseudocatalase is used topically on the lesional skin, and this is followed by UVB exposure to the whole body or to the lesional skin. The combination is claimed to correct the oxidative stress on melanocytes in vitiligo patients and thus lead to correction of the depigmentation.
Topical 5-Fluorouracil
Topical 5-fluorouracil is supposed to induce repigmentation of vitiligo lesions by overstimulation of follicular melanocytes which migrate to the epidermis during epithelialization.39 This form of topical therapy can be combined with spot dermabrasion of the vitiligo lesions to improve the repigmentation response. In a study by Sethi et al,40 a response rate of 73.3% was observed with a combination of spot dermabrasion and topical 5-fluorouracil after a treatment period of six months.40
Surgical therapies
Surgical therapies for vitiligo have further increased the percentage cure of the disease by an appreciable degree, with the consequent increase of their use in the management of unresponsive vitiligo both in India and abroad. These surgical therapies, as a rule, are indicated in those patients who have a stable (non-progressive) disease of at least one year and not responding to medical treatment. In general the most important advantage with these procedures is that the chances of repigmentation of lesions are in the range of 90-100%. Moreover, these interventions are becoming better and easier to perform with every passing day.
Different surgical therapies that have been attempted in the management of vitiligo include autologous suction blister grafting, split-thickness grafting, punch grafting, smash grafting, single follicular unit grafting, cultured epidermal suspensions and autologous melanocyte culture grafting. All these grafting procedures, except the melanocyte culture grafting, are easy to perform and do not require any sophisticated instruments. These grafting techniques have now been divided into two types, tissue grafts and cellular grafts, depending on whether whole epidermal/dermal tissue is transplanted or the individual cellular compartment.
Tissue grafting technique
Suction blister grafting
Here, thin epidermal grafts are taken from suction blisters on the donor site, usually on the buttocks or thighs. These suction blisters are produced by applying sufficient negative pressure on the skin at the donor site by using a suction apparatus or syringes with three-way cannulae. The epidermal grafts are then transplanted on to dermabraded vitiligo lesions. This leads to repigmentation of the recipient areas with an excellent cosmetic matching. The ease of the procedure, the high success rate and the excellent cosmetic results have all made suction blister grafting the procedure of choice in vitiligo grafting.41
Split thickness grafting
In this grafting technique a thin split thickness graft is taken from a donor site with the help of a dermatome, Humby’s knife, Silver’s knife or a simple shaving blade. This graft is then transplanted on to dermabraded recipient areas. This technique also gives excellent cosmetic matching after repigmentation and the incidence of repigmentation in this technique is also quite high. In fact, most comparison studies on grafting techniques in vitiligo have shown that maximum repigmentation is achieved with either suction blister grafting or split thickness grafting.41 The advantage of partial thickness grafting over the suction blister method is that a relatively larger area of vitiligo can be tackled in a single sitting. Both partial thickness skin grafting as well as suction blister grafting can be followed up by NB-UVB to achieve faster and better results.
Miniature punch grafting
Here full-thickness punch grafts of 1.0 to 2.0 mm diameter are taken from a suitable donor site and then transplanted on to similar punch shaped beds on the recipient vitiligo lesions. The recipient area is then treated with either PUVA/PUVA-sol or topical steroids leading to spread of pigment from the transplanted punches to the surrounding skin. With time the whole of the recipient area gets repigmented. The advantages of this procedure are that it is easy to perform and can take care of a relatively larger vitiligo area compared with the above two procedures. Also vitiligo lesions with irregular or geographical shapes can be treated with this procedure. However there are certain limitations. There is the risk of ‘cobblestone appearance’, ‘polka-dot appearance’, and hypertrophic changes at the recipient site.42 All these side effects can be minimized by proper patient selection and by use of smaller sized punches of 1.0 to 1.5 mm diameter. Miniature punch grafting is presently the commonest surgical procedure performed in India on vitiligo patients.
Follicular unit grafting
In this technique, single-hair follicular units are harvested/prepared from a suitable donor area as in the case of hair transplantation. These follicular units are then cut above the level of the follicular bulb and then transplanted into vitiligo lesions. The idea behind this technique is that the melanocytes in the follicular unit are ‘donated’ to the vitiliginous skin and serve as a source of pigment at the recipient site. The repigmentation process here simulates the normal process of repigmentation of vitiliginous skin quite closely and thus gives an excellent cosmetic result. This procedure combines the advantages of punch grafting with the excellent cosmetic results of split thickness or blister grafting techniques.43 The procedure is however tedious and needs good expertise on the part of the cosmetic surgeon.
Smash grafting
In this technique, a partial thickness graft is taken and is ‘smashed’, or cut into very small pieces, by means of a surgical blade on a suitable surface such as a glass slide. This ‘smashed’ tissue is then transplanted on to the dermabraded recipient skin and covered with a special powder or corrugated tube dressing so as to keep the smash-graft undisturbed on the recipient area. The advantage of this technique, over a simple partial thickness grafting, is that thicker grafts can be used with a good cosmetic result. The procedure has been indicated for those who are relatively inexperienced and cannot take an ideal, thin and transparent partial thickness graft from the donor area.44
Cellular grafting techniques
Non-cultured epidermal suspensions
Here a split-thickness graft is taken from a donor area and then incubated overnight. On the next day the cells are mechanically separated using trypsin-EDTA solution and then centrifuged to prepare a suspension. This cell suspension is then applied to the dermabraded vitiligo lesions, and a collagen dressing is applied to keep it in place. A relatively large area of vitiligo, about ten times the size of the donor graft can be taken care of with this procedure.45 The recipient area however has to be treated with either NB-UVB or PUVA for two to three months to achieve the desired pigmentation.
Melanocyte culture transplantation
This is a relatively more advanced grafting procedure where, once again, a split-thickness graft is taken from a donor area and incubated in an appropriate culture medium to grow the melanocytes or the keratinocytes-melanocyte combination in vitro. The cultured cells are then applied onto laser dermabraded, or even mechanically abraded, lesional skin.46,47 The procedure is obviously more difficult to perform, as it needs the advanced laboratory facilities for melanocyte culture. However the results with this procedure are excellent and a relatively large area of involved skin can be tackled by a single donor graft.
Summary
Table 2 summarises the above discussion of treatment options in vitiligo.
Table 2: New treatment options in vitiligo
Medical therapies and phototherapy
Surgical therapies
Narrowband UVB therapy either alone or in combination with immunomodulators, Vitamin D analogues etc.
Carers play a vital role in supporting family members who are sick, infirm or disabled.1 There is no doubt that the families of those with mental disorders are affected by the condition of their near ones. Families not only provide practical help and personal care but also give emotional support to their relative with a mental disorder. Therefore the affected person is dependent on the carer, and their well-being is directly related to the nature and quality of the care provided by the carer. These demands can bring significant levels of stress for the carer and can affect their overall quality of life including work, socializing and relationships. Research into the impact of care-giving shows that one-third to one-half of carers suffer significant psychological distress and experience higher rates of mental ill health than the general population. Being a carer can raise difficult personal issues about duty, responsibility, adequacy and guilt.2 Caring for a relative with a mental health problem is not a static process since the needs of the care recipient alter as their condition changes. The role of the carer can be more demanding and difficult if the care recipient’s mental disorder is associated with behavioural problems or physical disability. Over the past few decades, research into the impact of care-giving has led to an improved understanding of this subject including the interventions that help. It has now been realized that developing constructive working relationships with carers, and considering their needs, is an essential part of service provision for people with mental disorders who require and receive care from their relatives.
The aim of this review was to examine the relationship between caring, psychological distress, and the factors that help caregivers successfully manage their role.
‘Family burden’ - The role of families as carers
Caring for someone with a mental disorder can affect the dynamics of a family. It takes up most of the carers’ time and energy. The family’s responsibility in providing care for people with mental disorders has increased in the past three decades. This has been mainly due to a trend towards community care and the de-institutionalization of psychiatric patients.3 This shift has resulted in the transferral of the day-to-day care of people with mental disorders to family members. Up to 90% of people with mental disorders live with relatives who provide them with long-term practical and emotional support.4, 5 Carer burden increases with more patient contact and when patients live with their families.6 Strong associations have been noted between burden (especially isolation, disappointment and emotional involvement), caregivers’ perceived health and sense of coherence, adjusted for age and relationship.7
‘Family burden’ has been adopted to identify the objective and subjective difficulties experienced by relatives of people with long-term mental disorders.8 Objective burden relates to the practical problems experienced by relatives such as the disruption of family relationships, constraints in social, leisure and work activities, financial difficulties, and negative impact on their own physical health. Subjective burden describes the psychological reactions which relatives experience, e.g. a feeling of loss, sadness, anxiety and embarrassment in social situations, the stress of coping with disturbing behaviours, and the frustration caused by changing relationships.9 Grief may also be involved. This may be grief for the loss of the person’s former personality, achievements and contributions, as well as the loss of family lifestyle.10 This grief can lead to unconscious hostility and anger.9,10
The impact of caring on carers’ mental health
The vehicles of psychological stress have been conceptualized as adjustment to change,11 daily hassles,12 and role strains.13 Lazarus and Folkman (1984)14 define stress as ‘a particular relationship between the person and the environment that is appraised by the person as taxing or exceeding his or her resources and endangering his or her well being.’ The association between feelings of burden and the overall caregiver role is well documented.15 Caregivers provide assistance with activities of daily living, emotional support to the patient, and dealing with incontinence, feeding, and mobility. Due to high burden and responsibilities, caregivers experience poorer self-reported health, engage in fewer health promotion actions than non-caregivers, and report lower life satisfaction.16, 17
The overarching theme from the findings is that carers and care recipients do not believe that care recipients’ basic needs are being met, which causes them a great deal of distress and anger towards services and increases carer burden. Carers assert that the needs of care recipients and carers are interconnected and should not be seen as separate.18 The stress in carers is best understood by Pearlin`s stress-process model as shown in Figure 1.
Figure 1: Pearlin’s stress–process model of stress in carers (adapted from Pearlin et al, 1990)
The burden and depressive symptoms sustained by carers have been the two most widely studied care-giving outcomes. Reports indicate that depressive symptoms are twice as common among caregivers than non-caregivers.19 Family caregivers who have significantly depressed mood may be adversely affected in their ability to perform desirable health-maintenance or self-care behaviours in response to symptoms.20 Family caregivers experience more physical and mental distress than non-caregivers in the same age group.16 Several studies suggest that many caregivers are at risk of experiencing clinical depression.21 Nearly half of the caregivers in some studies were reported to meet the diagnostic criteria for depression when structured clinical interviews were used.22 There is also some evidence to suggest that a diagnosis of depression can be causally related to the care-giving situation. Dura et al (1991)23 found that nearly one quarter of caregivers met the criteria for depression whilst in the care-giving role, although they had never been diagnosed with depression prior to their assumption of this role. It has been proven that if the problem behaviours and the functional impairment in the care recipients is worse, the strain score is higher and the carer is more likely to be depressed.24 The societal implications of this are underscored by reports indicating that the stressed caregiver is more likely to institutionalize the care recipient.25, 26
The impact of caring for different mental disorders
The impact of caring for different mental disorders, and associated risk factors, is shown in table 1. Although only limited data is available on the psychological distress experienced by the carers of people with other mental disorders, it seems that these disorders have a significant impact on families. Obsessive-compulsive disorder has a considerable impact on families and can lead to a reduction in social activities, causing isolation over time.38 People with obsessive-compulsive symptoms frequently involve their relatives in rituals.38 This can lead to an increase in anger and criticism towards them which has a negative impact on treatment outcomes.38 Caring for patients with eating disorders can be overwhelming for the carer. Available data suggest that the impact on carers of persons with anorexia nervosa may be even higher than for psychoses.39 Studies on bulimia nervosa indicate that carers have significant emotional and practical needs.40
Table 1: The impact of caring for different mental disorders and associated risk factors
Mental Disorder
Risk factors
Impact on the carer
Schizophrenia28
High disability, very severe symptoms, poor support from professionals, poor support from social networks, less practical social support, violence.
Guilt, loss, helplessness, fear, vulnerability, cumulative feelings of defeat, anxiety, resentment, and anger are commonly reported by caregivers.
Dementia 29,30
Decline in cognitive and functional status, behavioural disturbances, dependency on assistance.31
Anger, grief, loneliness and resentment.
Mood disorders
Symptoms, changes in family roles, cyclic nature of bipolar disorder, moderate or severe distress.32
Significant distress,33 marked difficulties in maintaining social and leisure activities, decrease in total family income, considerable strains in marital relationships.34, 35
Psychological consequences during critical periods also persisting in the intervals between episodes
in bipolar disorder,36 poorer physical health, limited activity, and greater health service utilization than non-caregivers.37
Table 2: Risk factors for carer psychological distress
Caregiver factors
Research findings
Gender
·Women have higher rates of depression than men in the care-giving role.42
·39% of female caregivers, compared to 16% of male caregivers, qualified as being at-risk for clinical depression on The Center for Epidemiologic Studies-Depression Scale (CES-D).43
·A randomized controlled trial44 found that women were more likely than men to comply with a home environmental modification intervention, implement recommended strategies, and derive greater benefits.
·Male carers tend to have more of a ‘managerial’ style that allows them to distance themselves from the stressful situation to some degree by delegating tasks.45
Age
·Age-associated impairments in physical competence make the provision of care more difficult for older caregivers.
·There is a positive association of age and caregiver burden in Whites, but a negative association for African-Americans suggesting that older African-Americans are less likely to experience care-giving as physically burdensome.46
Caregiver health
·Caregiver health has also been identified as a significant predictor of caregiver depression.46
·Poorer physical health among caregivers than age-matched peers. Such health problems are linked to an increased risk of depression.47
·Longitudinal studies demonstrated that caregivers are at a greater risk, than non-care-giving age-matched controls, for developing mild hypertension and have an increased tendency to develop a serious illness48 as well as increased risk for all-cause mortality.49
Ethnicity
·Ethnicity has substantial impact on the care-giving experience.41
·Comprehensive reviews of the literature have identified differences in the stress process, psychological outcomes, and service utilization among caregivers of different racial and ethnic backgrounds.50
·Studies consistently show important differences in perceived burden and depression among African-American, White, and Hispanic family caregivers.51
·Caucasian caregivers tend to report greater depression and appraise care-giving as more stressful than African-American caregivers.52
·Hispanic caregivers report greater depression and behavioural burden than Caucasians and African-Americans.53
Social support
·Social support has profound effects on caregiver outcomes.
·More social support corresponds to less depressive symptomatology47 and lower perceived burden.54
·Care-giving is associated with a decline in social support, and increased isolation and withdrawal. 55
·Social support and caregiver burden have been found to mediate depression in caregivers.55
·Social support has other important functions in that carers may find out about services from people who have used them before and form a network with others in similar situations.41
Factors associated with psychological distress of the carer
Risks for carer psychological distress or depression are related to gender, age, health status, ethnic and cultural affiliation, lack of social support, as well as certain other characteristics related to the caregiver (table 2).41
Some of the patient factors related to psychological distress in carers are: behavioural disturbances, functional impairments, physical impairments, cognitive impairments, and fear that their relative may attempt suicide.
The frequency with which behavioural disturbances are manifested by the patient has been identified as the strongest predictor of caregiver distress and plays a significant role in the caregivers decision to institutionalize the patient.25 The literature consistently demonstrates that the frequency of behavioural problems is a more reliable predictor of caregiver burden and depression than are the functional and cognitive impairments of the individual.56 Carers face unfamiliar and unpredictable situations which increases stress and anxiety. Anxiety may be increased by behavioural problems of patients who cannot be successfully managed on a consistent basis.56 Anxiety is associated with depression, stress, and physical ill health.56
Findings regarding the relationship of functional impairment and negative caregiver outcomes have been inconclusive. Some studies document a weak association of objective measures of patient functional status and caregiver burden/depression,57 whereas others report a stronger relationship.54 Carers have reported great anxiety due to fear that their relative may attempt suicide.58 Carers of people with both physical and cognitive impairments have higher scores for objective burden of caring than those caring for people with either type of impairment alone. 58 In contrast, scores for limitations on their own lives were higher among women caring for people with cognitive impairments (with or without physical impairments).59
Coping styles and interventions to reduce psychological distress in carers
There is increasing interest in examining the factors that help caregivers successfully manage their role, while minimizing the effect on their mood and general well-being.60 Much of this research has been done within the general framework of stress and coping theory,61 examining coping styles of caregivers and the relationship between types of coping styles and reported symptoms of depression.62 A variety of interventions have been developed which support caregivers (table 3). Interventions include: training and education programs, information-technology based support, and formal approaches to planning care which take into account the specific needs of carers, sometimes using specially designated nurses or other members of the health care team.63
Ballard et al (1995)64 demonstrates that a higher level of carer education regarding dementia increases carers’ feelings of competency. This is more likely to reduce their expectations of their dependents’ abilities. Previous studies which have looked at these coping strategies and feelings of competence have shown that unrealistic expectations of a dependant increases carers’ risk of depression,65 and conversely a reduction of carers’ expectations is associated with lower rates of depression.66 Caregivers who maintain positive feelings towards their relative have a greater level of commitment to caring and a lower level of perceived strain.67 Furthermore, carers who experience feelings of powerlessness, lack of control, and unpreparedness have higher levels of depression.65 The most effective treatments in depression of carers appear to be a combination of education and emotional support.68
Spiritual support can also be considered a coping resource and has been studied in older African-Americans and older Mexican-Americans.69 Previous work examining the role of spiritual support observed that African-American caregivers report higher spiritual rewards for caregiving,70 and reliance on prayer and church support.71
Religious coping plays a paramount role, and it is often present at higher levels for African-Americans and Hispanics. For REACH caregivers, Coon et al (2004)72 found that religious coping is greater for Hispanic and African-American than for White caregivers. Religious involvement is frequently associated with more access to social support as well.73
Anecdotal literature74 suggests that caregivers who use more active coping strategies, such as problem solving, experience fewer symptoms of depression than do those who rely on more passive methods. Significant associations have been reported between positive strategies for managing disturbed behaviour, active strategies for managing the meaning of the illness, and reduced levels of caregiver depression. An important role for health-care professionals is in helping caregivers enhance their coping skills, supporting existing skills, and facilitating the development of new ones.66
Table 3: Coping styles and interventions to reduce psychological distress in carers
An important role for health-care professionals is in helping caregivers enhance their coping skills, supporting existing skills and facilitating the development of new ones.
· Training and education programs
· Information-technology based support
· Formal approaches to planning care
· Combination of education and emotional support
· Spiritual support
· Religious coping
· Positive strategies for managing disturbed behaviour
· High quality of informal relationships and presence of informal support
· Psychotherapy
· Cognitive-behavioural family intervention
Care-giving has some positive associations for caregivers, including pride in fulfilling spousal responsibilities, enhanced closeness with a care receiver, and satisfaction with one's competence.75 These perceived uplifts of care-giving are associated with lower levels of caregiver burden and depression.76 However perceived uplifts are more common among caregivers of colour than among Whites.77
High quality of informal relationships, and the presence of informal support, is related to lower caregiver depression78 and less deterioration in the emotional health for African-American caregivers, but not for Whites.79 Support of caregivers by others help to alleviate stress if the supporter is understanding and empathic.74 In one study, caring for a family member was not perceived to be a burden, and caregivers reported notable limitations on their social networks and social activities. They reported higher levels of unemployment than would be expected for the general population and were over-represented in lower income groups. Family carers are at high risk of social and economic disadvantage and at high risk of mental health challenges.80 Highly stressed persons may not be able to benefit from attempted social support of others as much as moderately stressed persons.81
Caregivers need to have the opportunity to learn more effective ways of coping with stress. If they can learn new ways to cope, they can reduce their anxiety and reliance on treatments.41 Bourgeois et al (1997)82 report that caregiver’s behavioural skills and effective self-management training programmes result in a lower frequency of patient behavioural problems and helps to improve the caregiver’s mood. Stevens and Burgio (2000)83 designed a caregiver intervention that teaches caregivers behavioural management skills to address problem behaviours exhibited by individuals with dementia, as well as problem-solving strategies to increase pleasant activities for the caregiver. Passive coping styles have been associated with greater burden. Persons who use an escape-avoidance type of coping are known to have more depression and interpersonal conflicts.41
Psychotherapy may be of some benefit in patients with early dementia but, due to cognitive loss, some adaptation of the technique is required and the involvement of carers is often necessary.84 Cognitive-behavioural family intervention can have significant benefits in carers of patients with dementia and has a positive impact on patient behaviour.85 From a cognitive perspective, care-giving plays an important invisible part, which consists of interpreting the care receiver's behaviour, reflecting on the best way to adjust to it, and defining care objectives.86 The interventions requiring active participation by the caregivers and those based on cognitive behavioural therapy can produce significant reductions in burden, anxiety and depression than those focused on knowledge acquisition.87
Among caregivers with depressive symptoms, 19% used antidepressants, 23% antianxiety drugs, and 2% sedative hypnotics. African-American caregivers were less likely than Whites to be taking antidepressants.88 In their study, Kales et al (2004)89 reported use of herbal products in 18% of elderly subjects with depression and/or dementia and in 16% of their caregivers.
In the Burdz et al (1988)90 study, respite care proved to have a positive effect on the burden experienced by the caregivers, and it also had a positive effect, against all expectations, on the cognitive and physical functioning of the persons with dementia.
There are more than twenty instruments that could be used as outcome measures with mental health carers and have good psychometric properties. They can measure (i) carers' well-being, (ii) the experience of care-giving and (iii) carers' needs for professional support.91 The caregiver burden scale and the sense of coherence scale seem to be highly useful for identifying carers at risk of stress, the pattern of burden, and coping strategies. Nurses can help family caregivers to identify their negative experiences about care-giving and can help them reflect upon their coping strategies to find balance in their situation. Risk groups of caregivers may be identified, especially those with a low perceived health and sense of coherence, for early interventions to reduce burden.7
Conclusion
The impact of caring for someone with mental illness brings the risks of mental ill health to the carer in the form of emotional stress, depressive symptoms, or clinical depression. Most individuals with mental disorders live in their own homes and are cared for by a family member. The caring process can be very taxing and exhausting, especially if the care recipient has a severe mental disorder. Providing such long-term care can be a source of significant stress. The behavioural problems associated with mental disorders further increase the stress levels of the carer and therefore impacts significantly on their mental health.
Carers face mental ill health as a direct consequence of their caring role and experience higher rates of mental ill health than the general population. This leads to negative effects on the quality of life of the carer and the standard of care delivered. Efforts to identify and treat caregiver psychological distress will need to be multidisciplinary, require consideration of the cultural context of the patient and caregiver, and focus on multiple risk factors simultaneously. The findings of the review underline the importance for early identification of carers, effective carer support, health promotion, monitoring high-risk groups, and timing appropriate interventions.
Acute Lung Injury (ALI) is a continuum of clinical and radiographic changes affecting the lungs, characterised by acute onset severe hypoxaemia, not related to left atrial hypertension, occurring at any age. At the severe end of this spectrum lies Acute Respiratory Distress Syndrome (ARDS) and therefore unless specifically mentioned this review will address ARDS within the syndrome of ALI.
It was first described by Ashbaugh in the Lancet in 1967. This landmark paper described a group of 12 patients with “Respiratory Distress Syndrome” who had refractory hypoxaemia, decreased lung compliance, diffuse infiltrates on chest radiography and required positive end expiratory pressure (PEEP) for ventilation.1
Key Points on Acute Lung Injury
Common, life threatening condition which is a continuum of respiratory dysfunction with ALI and ARDS being at either end of the spectrum
Risk factors include conditions causing direct and indirect lung injury, leading to an inflammatory response which can cause multiple organ failure
Damage to alveolar epithelial cells and capillary vasculature impair gas exchange and can lead to fibrosis
Management aims include supportive care, maintaining oxygenation and diagnosing and treating the underlying cause
Evidence supports low tidal volume ventilation and conservative fluid management
Long term outcomes relate to neuromuscular, neurocognitive and psychological problems rather than pulmonary dysfunction
This initial description gave only vague criteria for diagnosis, focused on the most severe end of the continuum and was not specific enough to exclude other conditions. A more precise definition was described by Murray et al. in 1988 using a 4 point lung injury scoring system including the level of PEEP used in ventilation, ratio of arterial oxygen tension to fraction of inspired oxygen (PaO₂/FiO₂), static lung compliance and chest radiography changes2. Despite being more specific and assessing severity it was too large and complex for practical purposes in the ICU setting.
It was not until 1994 that The American –European Consensus Conference on ARDS set the criteria used today to define both ALI and ARDS in research and clinical medicine. It recommended ALI be defined as “a syndrome of inflammation and increased permeability that is associated with a constellation of clinical, radiological and physiological abnormalities that cannot be explained by, but may coexist with, left atrial or pulmonary capillary hypertension” .3 They distinguished between ALI and ARDS based upon the degree of hypoxaemia present, as determined by the ratio of partial pressure of arterial oxygen to fractional inspired oxygen concentration (PaO₂/FiO₂), with ALI patients demonstrating a milder level of hypoxaemia. Additionally ARDS changed from Adult Respiratory Distress Syndrome to Acute Respiratory Distress Syndrome to account for its occurrence at all ages.
DIAGNOSIS AND PROBLEMS RELATED TO THIS
There are no gold standard radiological, laboratory or pathological tests to diagnosis ALI and ARDS and patients are given the diagnosis based on meeting the criteria agreed in 1994. (See Table 1)
ALI is diagnosed clinically and radiologically by the presence of non-cardiogenic pulmonary oedema and respiratory failure in the critically ill.
Table 1 – Diagnostic Criteria for ALI and ARDS
ALI
ARDS
Onset
Acute
Acute
Oxygenation (PaO₂/FiO₂) ratio in mmHg, regardless of ventilatory settings
<300
<200
Chest Radiological Appearance
Bilateral Pulmonary Infiltrations which may or may not be symmetrical
Bilateral Pulmonary Infiltrations which may or may not be symmetrical
Pulmonary Wedge Pressure
(in mmHg)
<18 or no clinical evidence of left atrial hypertension
<18 or no clinical evidence of left atrial hypertension
Meeting criteria, in itself, is not a problem when diagnosing conditions in the ICU setting, as sepsis and multi-organ failure are defined using consensus based syndrome definitions, however there are problems specifically related to ALI’s diagnosis.
In practice ALI and ARDS are clinically under-diagnosed, with reported rates ranging between 20 to 48% of actual cases.4 This is due to poor reliability of the criteria related to;
Non-specific radiological findings which are subject to inter-observer variability
Oxygenation criteria is independent of inspired oxygen concentration or ventilator settings including lung volumes and PEEP
Excluding cardiac causes of pulmonary oedema including left ventricular failure, mitral regurgitation and cardiogenic shock, in the ICU setting is difficult even when pulmonary artery catheters are used
The definition includes a heterogeneous population who behave very differently in response to treatment, duration of mechanical ventilation and severity of pulmonary dysfunction.
However this is the definition used by the ARDS network (a clinical network set up in 1994 by The National Heart, Lung and Blood Institute and the National Institutes of Health in the USA) for its clinical trials and on this basis it is validated.
EPIDEMIOLOGY
Incidence
Incidence of ALI is reported as 17-34 per 100,000 person years.5 Unfortunately despite population studies demonstrating fairly consistent trends regarding age (mean approximately 60years), mortality (35-40%) and ratio of ARDS to ALI (around 70%), incidence figures are less consistent internationally. A recent prospective population-based cohort study in a single US county demonstrated a higher incidence around 78.9 per 100,000 person years and inferred from this that 190,600 cases could occur in the USA alone each year.6 This variation is likely due to problems with reliability of diagnosis as illustrated above and also related to ALI generally presenting as a critical care illness making its epidemiology directly linked to availability of ICU resources.
Cases are only “captured” in the ICU setting and it potentially exists outside this environment in unknown quantities.7 Taking this into account means ALI and ARDS are probably far commoner in clinical practice than reported and many patients may meet the diagnosis yet be managed outside the ICU environment.8
Risk Factors
ALI is a multi-factorial process which occurs due to environmental triggers occurring in genetically predisposed individuals, as ALI-inducing events are common, yet only a fraction of those exposed develop the syndrome.
Environmental triggers for developing ALI can be divided into those causing direct and those causing indirect lung injury, with sepsis, either intrapulmonary or extrapulmonary being the commonest cause. (See table 2)
Table 2 Direct and Indirect triggers for ALI
Direct Lung Injury
Indirect Lung Injury
Common
Pneumonia
Aspiration of gastric contents
Less Common
Pulmonary contusion
Fat / Amniotic fluid embolism
High Altitude
Near Drowning
Inhalation Injury
Reperfusion Injury
Common
Sepsis
Severe trauma with shock and multiple transfusions
Less Common
Burns
Disseminated intravascular coagulation
Cardiopulmonary bypass
Drug overdose (heroin, barbiturates)
Acute pancreatitis
Transfusion of blood products
Hypoproteinaemia
At present there is research into the role of genetic factors and how they contribute to susceptibility and prognosis.9 It is difficult to assess the molecular basis of ALI due to the range of ALI inducing events which can cause the lung injury, the heterogeneous nature of the syndrome itself, presence of additional comorbidities, potentially incomplete gene penetrance and complex gene-environment interactions. However possible candidate genes which predispose patients to ALI have been identified and other genes exist which may influence its severity, thus providing targets for research in treatment development.
Secondary factors including chronic alcohol abuse, chronic lung disease and low serum pH may increase risk of developing ALI.⁷ There may be factors which are protective against its development, such as diabetes in septic shock patients,10 but further research is required.
PATHOPHYSIOLOGY
It is thought ALI patients follow a similar pathophysiological process independent of the aetiology. This occurs in two phases; acute and resolution, with a possible third fibrotic phase occurring in a proportion of patients.
Acute Phase
This is characterised by alveolar flooding with protein rich fluid secondary to a loss of integrity of the normal alveolar capillary base, with a heterogeneous pattern of alveolar involvement.
There are two types of alveolar epithelial cells (Table 3), both of which are damaged in ALI, likely via neutrophil mediation, with macrophages secreting pro-inflammatory cytokines, oxidants, proteases, leucotrienes and platelet activating factor.
Table 3 Characteristics of Type I and Type II Alveolar Epithelial Cells
Type I
Type II
Percentage of cells
90%
10%
Shape
Flat
Cuboidal
Function
Provide lining for alveoli
Replace damaged type I cells by differentiation
Produce surfactant
Transport ions and fluids
Damage to type I alveolar epithelial cells causes disruption to alveolar-capillary barrier integrity and allows lung interstitial fluid, proteins, neutrophils, red blood cells and fibroblasts to leak into the alveoli.
Damage to type II cells decreases surfactant production and that produced is of low quality, likely to be inactivated by fluid now in alveoli, which leads to atelectasis. Additionally there is impaired replacement of type I alveolar epithelial cells and an inability to transport ions and therefore remove fluid from the alveoli.
Coagulation abnormalities occur including abnormal fibrinolysis and formation of platelet and fibrin rich thrombi which result in microvascular occlusion, causing intrapulmonary shunting leading to hypoxaemia.
Ventilation-perfusion mismatch, secondary to alveolar collapse and flooding, decreases the number of individual alveoli ventilated, which in turn increases alveolar dead space, leading to hypercapnia and respiratory acidosis. Additionally pulmonary compliance decreases and patients start to hyperventilate in an attempt to compensate the above changes.
The release of inflammatory mediators from damaged lung tissue triggers systemic inflammation and systemic inflammatory response syndrome (SIRS) which may progress to multiple organ failure, a leading cause of death in ARDS patients.
Resolution Phase
This phase is dependent on repair of alveolar epithelium and clearance of pulmonary oedema and removal of proteins from alveolar space.
The type II alveolar epithelial cells proliferate across the alveolar basement membrane and then differentiate into type I cells. Fluid is removed by initial movement of sodium ions out of the alveoli via active transport in type II alveolar epithelial cells, with water then following, down a concentration gradient through channels in the type I alveolar epithelial cells.
Soluble proteins are removed by diffusion and non soluble proteins by endocytosis and transcytosis of type I alveolar epithelial cells and phagocytosis by macrophages.
Fibrotic Phase
Some patients do not undergo the resolution phase but progress to fibrosing alveolitis, with fibrosis being present at autopsy in 55% non-survivors of ARDS.11 This occurs by the alveolar spaces filling with inflammatory cells, blood vessels and abnormal and excessive deposition of extracellular matrix proteins especially collagen fibres.12 Interstitial and alveolar fibrosis develops, with an associated decrease in pulmonary compliance and only partial resolution of pulmonary oedema with continued hypoxaemia.
CLINICAL FEATURES
Acute Phase
The diagnosis should be considered in all patients with risk factors who present with respiratory failure, as the onset though usually over 12 to 72 hours, can be as rapid as 6 hours in presence of sepsis.
Patients present with acute respiratory failure where hypoxaemia is resistant to oxygen therapy and chest auscultation reveals diffuse, fine crepitations, indistinguishable from pulmonary oedema.
Resolution Phase
This phase usually occurs after around 7 days after onset of ALI, where a resolution of hypoxaemia and improvement in lung compliance is seen.
Fibrotic Phase
There is persistent impairment of gas exchange and decreased compliance. In severe cases it can progress to pulmonary hypertension through damage to pulmonary capillaries and even severe right heart failure, with the signs and symptoms of this developing over time.
INVESTIGATIONS
Diagnostic criteria require arterial blood gas analysis to demonstrate the required ratio between the partial pressure of arterial oxygen and fractional inspired oxygen concentration.
Radiological Findings
Although there are no pathognomonic radiographic findings for ALI, features on plain chest radiography include;
Bilateral patchy consolidation, which may or may not be symmetrical
Normal vascular pedical width
Air bronchograms
Pleural effusion may be present
10-15% patients have pneumothoraces independent of ventilator settings
Computer tomography of the chest can show the heterogeneous nature of ALI, with dependent areas of the lung showing patchy consolidation with air bronchograms, atelectasis and fibrosis. As with plain radiography there may be pneumothoraces present.
Computer tomography and Chest radiograph of ARDS
MANAGEMENT
The aims of management are to provide good supportive care, maintain oxygenation and to diagnose and treat the underlying cause.
General
Good supportive care, as for all ICU patients, should include nutritional support with an aim for early enteral feeding, good glycaemic control and deep venous thrombosis and stress ulceration prophylaxis. It is important to identify and treat any underlying infections with antibiotics targeted at culture sensitivities and if unavailable, towards common organisms specific to infection site.
It is not uncommon for ALI patients to die from uncontrolled infection rather than primary respiratory failure.
Ventilator associated pneumonia is common in patients with ALI and can be difficult to diagnoses, as ALI radiological findings can mask new consolidation and raised white cell count and pyrexia may already be present. If suspected this should be treated with appropriate antibiotics, although long term ventilation can cause colonisation which leads to endotracheal aspirate culture results being difficult to interpret.
Although the role of physiotherapy in ALI is unclear, aims of treatment should be similar to those in all ICU patients, including removal of retained secretions and encouragement of active and passive movements, as patients are often bed bound for prolonged periods of time.
Ventilation
MODE OF VENTILATION
Ventilation is usually via endotracheal intubation using intermittent positive pressure ventilation with PEEP. There may be a role for non invasive ventilation in early stages of ALI, but it is poorly tolerated at higher PEEP settings which may be required to maintain oxygenation, and no evidence supports its use at present. Additionally there is no evidence to suggest an advantage of either volume or pressure controlled ventilation.
Principles of ventilation in ALI are to maintain adequate gas exchange until cell damage resolves whilst avoiding ventilator associated injury from;
Barotrauma – alveolar overdistension associated with ventilation at high volumes
Volutrauma – alveolar overdistension associated with ventilator high pressures
Biotrauma – repeated opening and closing of collapsed alveoli causing shearing stress which can initiate a proinflammatory process
Lungs in patients with ALI are heterogeneous and therefore can react variably to changes in ventilator settings. Therefore settings which provide adequate oxygenation, may damage more “healthy” areas of lung.13
Table 4 Lung ventilation in different parts of lung with acute lung injury
Area
Characteristics
Behaviour when ventilated
1
Normal compliance and gas exchange
Easily over ventilated
Exposed to potential damage
2
Alveolar flooding and atelectasis
Alveoli can still be recruited for gas exchange by safely raising airway pressures
3
Severe alveolar flooding and inflammation
Alveoli cannot be recruited without using unsafe airway pressures
TIDAL VOLUMES
Old strategies of high volume ventilation are likely to over inflate healthy lung portions leading to barotrauma and ventilator management in ALI has moved towards lower tidal volumes. This is a consequence of the ARDSnet tidal volume study, which demonstrated significant reduction in mortality (40 to 31%) when using a low volume ventilator strategy based on predicted body weight (6mls/kg and peak pressures <30cmH₂O vs. 12mls/kg and peak pressures <50cmH₂O).14 Furthermore they showed a decrease in systemic inflammatory markers, lower incidence of multiple organ failure and an increase in ventilator free days in the lower tidal volume group.
PEEP
It was postulated that PEEP may be beneficial in ARDS as it reduces biotrauma, maintains the patency of injured alveoli, reduces intrapulmonary shunting and improves ventilation-perfusion mismatch. However evidence regarding its use is inconclusive. Numerous large centre trials have demonstrated no difference in outcome or mortality between patients ventilated with lower PEEP vs. higher PEEP (8 vs. 14 cm H₂O).151617 Yet a recent JAMA systematic review and meta-analysis showed that although higher PEEP ventilation was not associated with improved hospital survival, it was associated with improved survival among the ARDS subgroup of ALI and suggested that an optimal level of PEEP remains unestablished but may be beneficial.18
ECMO (Extracorporeal membrane oxygenation)
This is a modified longer term form of cardiopulmonary bypass which aims to provide gas exchange across an artificial membrane external to the body, allowing the lungs time to recover. It is confined to a few specialist centres in the UK and the first results from the CESAR multicentre randomised controlled trial were published in the Lancet in 2009. It showed improved survival in adult patients with severe but potentially reversible respiratory failure on ECMO, as compared to conventional ventilation and demonstrated cost effectiveness in settings like the UK healthcare system.19 This therefore may be a treatment strategy to consider in extreme cases resistant to conventional therapy.
OTHER STRATEGIES
A current meta-analysis looking at prone positioning concluded that randomised controlled trials failed to demonstrate improved outcomes in ARDS patients overall. There is a decrease in absolute mortality in severely hypoxaemic patients with ARDS but as long term proning can expose ALI patients to unnecessary complications, it should only be used as rescue therapy for individuals resistant to conventional treatment.20
No evidence supporting specific weaning programmes exists and a recent Cochrane review showed no evidence to support recruitment manoeuvres in ALI. 21
Therefore the aim of ventilation is low volumes with permissive hypercapnia, providing adequate oxygenation (regarded as a partial pressure of arterial oxygen >8kPa) whilst trying to avoid oxygen toxicity lung injury.
Fluid Management
Fluid management has to balance the need for enough fluid to maintain an adequate cardiac output and end organ perfusion, with a low enough intravascular pressure to prevent high capillary hydrostatic pressures, which could cause pulmonary oedema, worsen oxygen uptake and carbon dioxide excretion. Evidence supports a negative fluid balance in patients not requiring fluid for shock.
Studies as early as 1990 showed a reduction in pulmonary wedge pressure was associated with increased survival22 and extravascular lung water was associated with poor outcomes23 in ARDS patients.
The ARDSnet FACTT study looked at two fluid regimens comparing liberal fluid management (a net gain of approximately 1 litre per day) with a conservative fluid management (zero net gain over first seven days).24 Although there was no significant difference in (the) primary outcome of 60 day mortality, the conservative management group had improved lung function, shortened duration of mechanical ventilation and intensive care and had no increased incidence of shock or use of renal replacement. This is supported by a recent retrospective review, which concluded negative cumulative fluid balance at day 4 of acute lung injury is associated with significantly lower mortality, independent of other measures of severity of illness.25
Pharmacotherapy
To date no pharmacological agent has been demonstrated to reduce mortality among patients with ALI.26 However ALI encompasses a wide range of patients with varying aetiology and comorbidities. It may be that on subdividing ALI patients, some therapies may be suitable for specific circumstances but at present there is little literature to support this.
EXOGENOUS SURFACTANT
Since the 1980’s numerous randomised controlled trials have demonstrated no benefit from synthetic, natural or recombinant surfactant use in adults with ALI.
INHALED NITRIC OXIDE
Despite providing selective vasodilatation and improving ventilation perfusion mismatch, trials have only showed short lived improvement in oxygenation and no change in mortality with nitric oxide use. At present it plays no role in standard ALI treatment and should be reserved for rescue therapy in patients difficult to oxygenate.27
STEROIDS
Despite the potential for steroids to benefit ALI patients due to anti-inflammatory properties, clinical trials demonstrate no improved mortality when given early or late in disease progression and given concerns regarding their role in development of neuromuscular disorders associated with critical illness, a recent large randomised controlled trial argued against steroid use in ALI.28
INTRAVENOUS SALBUTAMOL
Beta 2 agonists were shown to be experimentally beneficial in ALI due to increasing fluid clearance from alveolar space, anti-inflammatory properties and bronchodilation.29 The BALTI trial published in 2006, investigated the effects of intravenous salbutamol in patients with ARDS. It showed decreased lung water at day 7, lowered Murray lung injury scores and lower end expiratory plateau pressures but an increase in incidence of supraventricular tachycardias and therefore further investigation is needed before it can be recommended as treatment for ALI.30 The BALTI-2 trial is currently underway in the UK, to further assess possible benefits and complications.
Other new and promising treatments which are currently being evaluated in trials are activated protein C and granulocyte-macrophage colony-stimulating factor (GM-CSF).
MORTALITY
Mortality rates of patients with ALI and ARDS are similar, with both being around 35-40%.³ Controversy exists regarding whether mortality rates in ALI are decreasing,31 or have stayed static.32 Nonetheless death in patients with ALI is rarely from unsupportable hypoxaemic respiratory failure but from complications of the underlying predisposing conditions or multiple organ failure.33
There is some evidence related to racial and gender differences in mortality (worse in African Americans and males)34 and that thin patients have increased mortality and obese patients have somewhat lower mortality than normal weight individuals35 but the main independent risk factors for increased mortality are shown in Table 5.
Table 5 Independent risk factors for increased mortality in ALI as identified in multicentre epidemiological cohorts
·Old age
·Worse physiological severity of illness
·Shock, on admission to hospital
·Shorter stay in the ICU after ALI onset
·Longer hospital stay before ALI onset
·Increased opacity on chest radiography
·Immunosupression
OUTCOMES
Long term problems are related to neuromuscular, neurocognitive and psychological dysfunction rather than pulmonary dysfunction. (Table 6) There is poor understanding of the mechanisms which cause these sequelae and therefore prevention of these outcomes and planning rehabilitation can be difficult.
Table 6 Long Term Outcomes in ARDS survivors and caregivers
Neuromuscular dysfunction
·critical illness polyneuropathy
·critical illness myopathy
·entrapment neuropathy
Neurocognitive dysfunction involving
·memory
·executive function
·attention
·concentration
Psychological dysfunction
·Post traumatic stress disorder
·Depression
·Anxiety
Other
·Pulmonary dysfunction
·Tracheostomy site complications
·Striae
·Frozen joints
Caregiver and financial burden
A recent study into patients who survived ALI showed they require support during discharge from ICU to other hospital settings and again once in the community regarding guidance on home care, secondary prevention and support groups.36
CONCLUSION
The syndrome which encompasses ALI and ARDS is common and under-recognised, with many clinicians encountering it outside the ICU setting. Despite advances in identification and management, morbidity and mortality is still high. Care should focus on supportive treatment and managing the underlying cause, whilst specifically aiming for low volume ventilation and conservative fluid balance. Ongoing research is still needed to hone the diagnostic criteria, define genetic risk factors and develop new treatment strategies to improve outcome. The new challenge for clinicians is how to address the long term outcomes of survivors and their relatives which will be an increasingly important problem in the future.²⁶
Forty one years after the last influenza pandemic, while everyone was worrying about the avian influenza A (H5N1) virus causing a pandemic, an apparent new chapter is opened with the emergence of new strain of influenza A virus. On 24th April, the World Health Organization (WHO) declared the first ever public health emergency of international concern indicating the occurrence of confirmed human cases of swine influenza in Mexico and United States.1 Subsequently the Centre for Disease Control and Prevention (CDC) confirmed that these human influenza cases were caused by a novel strain of influenza A virus to which there is little or no population immunity.2 On June 2009, the WHO rated the pandemic alert from phase 5 to 6, signalling that the first pandemic of the 21st century was underway. It was however stressed that the rise in the pandemic alert level was mainly attributed to the global spread of the virus rather than its severity. The pandemic potential of influenza A viruses has been ascribed to their genetic and antigenic instability and there ability to transform by constant genetic re-assortment or mutations, which can result in the emergence of novel progeny subtypes capable of both infecting and leading to sustained person to person transmission.3 The newly emerged strain contains a combination of gene segments that have not been previously identified in swine or human influenza viruses.4
Historical Perspectives
Influenza has been recognised for hundreds of years, but the cause was unknown for most of this time. Hippocrates had defined this disease about 2400 years ago, but lacked laboratory confirmation.5 The year 1580, marks the first instance of influenza recorded as an epidemic even though there is possibility that there were many prior influenza epidemics.6 The word influenza (meaning influence), first used in 1743 originated from the Latin word “Influenza”, named so because the disease was considered to be caused by unfavourable astrological conditions. Since 1700, there have been approximately a dozen influenza A virus pandemics and the lethal outbreak of 1918-1919 is dubbed as the greatest medical holocaust in recorded history, killing up to 50 million people worldwide.7
The earliest evidence of influenza A virus causing acute respiratory illness in pigs was traced to the 1930s. Swine influenza A viruses are antigenically very similar to the 1918 human influenza A virus and they may all have originated from common ancestor.8 From 1930 to 1990, classic swine influenza A was the commonest swine influenza virus circulating amongst the swine population during which the virus did not undergo much genetic change. Antigenic variants of these classical influenza viruses emerged in 1991 and the real antigenic shift occurred at the ends of last century when the classical swine influenza virus re-assorted with human influenza A virus and a North American lineage avian influenza virus. This resulted in the emergence of multiple subtypes including H1N2 and H3N2. In the past few years, sporadic cases of human infections caused by swine influenza A virus have occurred, mainly due to subtypes. Occupational exposure to swine was the most important risk factor for infection and fortunately all patients recovered without resulting in efficient, sustained human to human transmission.9
Origin of 2009 Strain
The pandemic that began in March 2009, was originally referred to as “swine flu” because laboratory testing showed that many of the genes in this new virus were very similar to influenza viruses that normally occur in pigs (swine) in North America. But further study has shown that this new strain of virus represents a quadruple re-assortment of two swine strains, one human strain and one avian strain of influenza. The largest proportion of genes come from swine influenza viruses (30.6% from North American swine influenza strains, and 17.5% from Eurasian swine influenza strains), followed by North American avian influenza strains (34.4%) and human influenza strains (17.5%).10 Analysis of the antigenic and genetic characteristics of the pandemic influenza A virus demonstrated that it’s gene segments have been circulating for many years, suggesting that lack of surveillance in swine is the reason that this strain had not been recognized previously.11 This novel strain is antigenically distinct from seasonal influenza A and possesses previously unrecognised molecular determinants that could be responsible for the rapid human to human transmission. Moreover, antigenic drift has occurred amongst different lineages of viruses, therefore, cross protection antibodies against avian, swine and human viruses are not expected to exist. Emerging scientific data support the hypothesis of a natural genesis, with domestic pigs a central role in the generation and maintenance of the virus. Protein homology analysis of more than 400 protein sequences from the new influenza virus as well as other homologous proteins from influenza viruses of the past few seasons also confirmed that this virus has a swine lineage.1 Phylogenetic analysis has suggested that initial transmission to humans occurred several months before the recognition of the outbreak and multiple genetic ancestry of this influenza A is not indicative of artificial origin.11
Situation Update
In March 2009, an outbreak of respiratory illness was first noted in Mexico, which was eventually identified as being related to influenza A.12 The outbreak spread rapidly to the United States, Canada and throughout the world as a result of airline travel.13 On 11th June 2009, the WHO raised its pandemic alert to the highest level i.e. phase 6, indicating widespread community transmission on at least two continents.14
Pandemic influenza was the predominant influenza virus circulating in the US, Europe, northern and eastern Africa and in Australia. Activity of the virus has initially peaked and then declined in North America and in parts of western, northern and Eastern Europe, but activity continued to increase in parts of central and southeastern Europe, as well as in central and south Asia. As of 28th February 2010, worldwide more than 213 countries and overseas territories or communities have reported laboratory confirmed cases of pandemic influenza 2009, including at least 16455 deaths; a number the WHO acknowledges significantly underreported the actual number.15 Most of the deaths have been related to respiratory failure resulting from severe pneumonia and acute respiratory distress syndrome.16
In India, the number of confirmed cases till March 2010 was 29,953 and a total of 1410 deaths were reported. The rate of infection has been highest among children and young individuals of <24 years of age. To date, pandemic influenza A infections are uncommon in persons older than 65 years, possibly as a result of pre-existing immunity against antigenically similar influenza viruses that circulated prior to 1957.17 High rates of morbidity and mortality has been noted among children and young adults with underlying health problems including chronic lung disease, immunosuppressive conditions, cardiac disease, pregnancy, diabetes mellitus and obesity.18
Transmission and Shedding
Novel virus is contagious and can transmit from human to human in ways similar to other influenza viruses. The main route of transmission between humans is via inhalation of infected respiratory droplets (range in size from 0.08 µm to 0.12 µm) produced after coughing and sneezing.19 Transmission via contact with surfaces that have been contaminated with respiratory droplets or by aerosolised small-particle droplets may also occur. In addition to respiratory secretions, all other body fluids (including diarrhoeal stool) should also be considered potentially infectious.
The estimated incubation period is unknown and could range from 1 to 7 days, although the median incubation period in most cases appears to be approximately 2 days.20 Shedding of the virus begins the day prior to the onset of symptoms and can persist for 5-7 days in immunocompetent individuals. The amount of virus shed is greatest during the first 2-3 days of illness. Persons who continue to be ill, for a period of longer than 7 days after illness onset, should be considered potentially contagious until symptoms have resolved. Longer periods of shedding may occur in children (especially young infants), elderly adults, and patients with chronic illnesses and immunocompromised hosts who might be contagious for longer periods.
Clinical Manifestations
According to the CDC, in humans the symptoms of the 2009 “flu” virus are similar to those of influenza and of influenza-like illness in general. The illness with the virus has ranged from mild to severe and symptoms include fever, cough, sore throat, body aches, headache, chills and fatigue, which are usual features of influenza virus. The 2009 outbreak has shown an increase percentage of patients reporting diarrhoea and vomiting.16 As these symptoms are not specific to swine flu hence a differential diagnosis of probable swine flu requires not only symptoms but also a high likelihood of swine flu due to person’s recent history. The CDC advised physicians to consider swine influenza infection in the differential diagnosis of patients with acute febrile respiratory illness who have either been in contact with persons with confirmed swine flu or who were in states that have reported swine flu cases during the 7 days preceding their illness onset.
The overall severity with this 2009 virus has been less than what was observed during the influenza pandemic of 1918-1919. Most patients appear to have uncomplicated, typical influenza-like illness and recovered without requiring any medical treatment. About 70% of people who have been hospitalised have had one or more medical conditions, which include pregnancy, diabetes, heart disease, asthma and kidney disease.21 The most common cause of death is acute respiratory distress syndrome. The other causes of death are severe pneumonia with multifocal infiltrates (leading to sepsis), high fever (leading to neurological problems), dehydration (from excessive vomiting and diarrhoea) and electrolyte imbalance. Fatalities are more likely in young children (<5 years), elderly (>65 years) and in people with underlying conditions, which include pregnancy, asthma, lung diseases, diabetes, morbid obesity, autoimmune disorders, immunosuppressive therapies, neurological disorders and cardiovascular disease.22
Laboratory Diagnosis
All diagnostic laboratory work on clinical samples from suspected cases of virus infection should be done in a Biosafety Level 2 (BSL-2) Laboratory. Suspected cases of novel infection should have respiratory specimens (nasopharyngeal, nasal or oropharyngeal swab, bronchoalveolar lavage and endotracheal aspirate) collected to test for the 2009 flu virus. Specimens should be placed into sterile viral transport media (VTM) and to be kept at 4°C. Real time reverse transcriptase polymerase chain reaction (RT-PCR) is the recommended sensitive method for the detection of virus, as well as to differentiate between pandemic 2009 and regular seasonal flu.23 The other rapid influenza diagnostic tests (RIDTs), although provide results within 30 minutes or even less, none of these tests can distinguish between influenza A virus subtypes. Moreover, RIDTs do not provide any information about antiviral drug susceptibility. Isolation of the virus in cell cultures or embryonated eggs is another method for diagnosis of infection, but may not yield timely results for clinical management and negative viral culture does not exclude the influenza A infection.
However, most people with flu symptoms do not need a test for pandemic 2009 flu, specifically because the test results usually do not affect the recommended course of treatment. The CDC recommends testing only for people who are hospitalised with suspected flu and persons having underlying medical conditions and those with weak immune systems.24 It is also expressed that treatment should not be delayed by waiting for laboratory confirmation of test results, but rather make diagnosis based on clinical and epidemiological backgrounds and start treatment early.
Treatment
The virus isolates in the 2009 outbreak are found to be resistant to amantidine and rimantidine. The CDC recommends the use of neuraminidase inhibitors as the drugs of choice for treatment and prevention of 2009 influenza in both children and adults.25 Tamiflu (oseltamivir phosphate) and Relenza (zanamivir) are the two FDA-approved influenza antiviral drugs and a third neuraminidase inhibitor peramivir is an experimental drug approved for hospitalised patients in cases where the other available methods of treatment are ineffective or unavailable. Antiviral drugs not only make the illness milder but also prevent serious flu complications. However, the majority of people infected with the virus make a full recovery without requiring medical attention or antiviral drugs. Treatment is recommended for patients with confirmed or suspected 2009 influenza who have severe, complicated or progressive illness or who are hospitalised. People who are not from the at-risk group and have persistent or rapidly worsening symptoms should also be treated with antivirals. Therapy should be started as soon as possible, since evidence of benefit is strongest when treatment is started within 48 hours of illness onset.26 Treatment should not be delayed while awaiting the results of diagnostic testing nor should it be withheld in patients with indications for therapy who present >48 hours after the onset of symptoms. Beside antivirals, supportive care at home or in hospital, focuses on controlling fevers, relieving pain and maintaining fluid balance as well as identifying and treating any secondary infections or other medical problems.
Major Concern
The neuraminidase inhibitors oseltamivir and zanamivir provide valuable defences and have been used widely for treatment and chemoprophylaxis of 2009 pandemic influenza A.But the recent emergence of resistance to these antiviral drugs is a matter of immediate concern. Influenza A strain resistant to oseltamivir has been reported from a variety of geographical locales and poses a challenge for the management of severely compromised patients.27 The CDC warned that the indiscriminate use of antiviral medications to prevent and treat influenza could ease the way for drug resistant strains to emerge, which would make the fight against the pandemic much harder. Most of the patients recover spontaneously without any medical attention and use of antiviral medications should be reserved primarily for people hospitalised with pandemic flu and persons, with pre-existing or underlying medical conditions who are at higher risk for influenza-related complications. It has also been emphasised that early treatment once a patient has developed symptoms, rather than chemoprophylaxis, should reduce opportunities for the development of oseltamivir resistance.26 The degree to which these drugs will remain effective for the treatment of the novel strain of influenza in the coming months is still a question.
What’s next?
The only possible way to combat the situation is large scale immunization. Antiviral drugs are not a substitute for vaccinination and are used only as an adjunct to vaccines in the control of influenza. Vaccines are one of the most effective ways to protect people from contracting illness during epidemics and pandemics of influenza. The seasonal vaccines do not confer any protection against 2009 H1N1; new vaccines have been licensed and are available.28 The vaccines are available in both live-attenuated and inactivated formulations. Two types of vaccines are approved by the FDA for use in the prevention of 2009 pandemic influenza virus. These are TIV (“flu shot” of trivalent inactivated vaccine) and LAIV (nasal spray of live attenuated vaccine). The inactivated vaccine is contraindicated in patients with severe allergic reaction to eggs or any other component of the vaccine. The live attenuated vaccine is licensed for persons aged 2 through 49 years who are not pregnant, are not immunocompromised and have no underlying medical conditions. Children less than 5 years who have asthma and are taking long term aspirin therapy should also not receive live vaccines. Otherwise, both vaccines are safe and highly immunogenic and a single administration leads to robust immune response in 80% to 90% of adults aged 18-64 years and in 56% to 80% of adults aged 65 years and older with in about 10 days.29 Children younger than 10 years will require two administrations of the vaccine separated by at least 21 days. Adverse effects following vaccination are minor, just like those of seasonal influenza vaccine and are self limiting. Concerns regarding the risk of Guillain-Barre syndrome (GBS) after vaccination have been raised. Various studies have suggested that the risk of GBS is higher from influenza itself rather than from the vaccine and the other adverse effects.30 The CDC is now encouraging everyone including people of 65 years and above to get vaccinated against the 2009 strain of influenza.
The Government of India has recently approved a split virus, inactivated, non-adjuvant monovalent vaccine (Panenza by Sanofi Pasteur) to inoculate frontline health workers and those who have a high risk of getting infected.31 Groups of health care workers has also been singled out by the European council for attention and immunization.32 Infection control practices in the health care settings should be followed along with as per the guidelines.33 Patients should also be educated regarding the other preventive measures, including using tissues to cover their mouth and nose when coughing and sneezing, developing good hand washing techniques, use of alcohol based hand-rubs, avoiding contact with ill persons if possible and staying home when ill unless medical attention has been given.
The flu season seems to be dying down in 2010 but the war is yet not over. Lessons must be learnt from the previous influenza pandemics and it is still important to get vaccinated against the flu and be prepared, as activity as well as virulence might increase again in the coming season. The words of Margaret Chan (Director General, WHO) to be remembered that “the virus writes the rule and this one like all influenza viruses can change the rules, without rhyme or reason, at any time”.
Female sexual dysfunction (FSD) is a serious morbidity which could occur postnatally. It may lead to a variety of physical, psychological, and social adverse effects on the patient. Moreover, the consequent cycle of fear might compound the initial sexual disorder and makes it more difficult to treat. Therefore, early diagnosis and management of the problem become essential to avoid later sequalae on reproductive and sexual life. However, early diagnosis may be challenged by many factors. For example, many patients will be preoccupied by the newborn or embarrassed of talking about sexual matters after delivery, which makes it very important for the midwifery, medical, or other staff to raise the issue during the postnatal care sessions. The staff, on the other hand, might feel uncomfortable to discuss the sexual function with the client, or even may lack the knowledge and skills required for sexual health counselling. In addition to the client-service gap, there are gaps between different sexual service providers.
There are many types of postnatal sexual disorders. These types can differ widely in clinical features and management. Additionally, management of postpartum female sexual dysfunction (PPFSD) can vary with clinician’s experience. There are very few randomised clinical trials on treatment for PPFSD, which partly explains the service-service gap in PPFSD management.
In the last three decades there has been an increase in caesarean section rate in the developed world due to many maternal and fetal indications, especially with the significant improvement in surgical and postoperative care. Recently, more attention has been paid to the positive role the caesarean section may play in protecting the female pelvic floor from birth trauma. Perineal birth trauma has been accused by many authors of adversely affecting the female sexual well being. 1 On the other hand there is a growing opinion that the quality of postnatal sexual health is unrelated to mode of delivery. 2 The previous two contradictory statements from literature illustrate the real size of the dilemma when we try to counsel a woman requesting a caesarean section as she is worried about sexual dysfunction after vaginal delivery. This problem might become more difficult to solve if the woman already suffers from a sexual disorder (for example: dyspareunia) in the antenatal or preconception period.
Female sexual dysfunction is impaired or inadequate ability of a woman to engage in or enjoy satisfactory sexual intercourse and orgasm. There are certain natural events in a woman’s life when she is at increased risk of developing sexual dysfunction, such as the use of contraception pills, menstruation, postpartum and lactation status, perimenopause, and postmenopause. This could be related to fluctuations in gonadal hormone secretion, making women more vulnerable to sexual symptoms.3 Postpartum female sexual dysfunction (PPFSD) is a common health problem with different incidence reported in literature. Xu et al reported an incidence of 70.6% of PPSFD in the first 3 months after delivery falling off to 55.6% during the 4th-6th months, and reduced to 34.2% at the 6th month, but not reaching pre-pregnancy levels of 7.17%. 2
For the purpose of this piece of writing, the classification of sexual dysfunction put forth by the American Psychiatric Association APA (1994) in the Diagnostic and Statistical Manual, 4th Edition (DSM-IV) is used to help understand the differing presentations of PPFSD. 4 The main postpartum female sexual dysfunction categories are: sexual desire dysfunction (Hypoactive Sexual Desire Disorder), sexual pain disorders (which includes dyspareunia, vaginismus. and vulvodynia), sexual arousal disorder, and female orgasmic disorder.
To help in understanding this classification better, it is important to refer to the early research done in this field by Masters and Johnson in 1966. One of the most interesting findings of the latter has been the four stage model of sexual response, which they described as the human sexual response cycle.5 They divided the human sexual response cycle into four stages: Excitement phase (initial arousal), Plateau phase (at full arousal, but not yet at orgasm), Orgasm, and Resolution phase (after orgasm). 5
Although it is normal to have hypoactive sexual desire (loss of libido) in the first 6-7 weeks after giving birth, this becomes abnormal when the desire for sexual activity is persistently reduced or absent causing distress in the relationship. Sexual desire disorder after delivery may be due to the mother being preoccupied with the neonate or postpartum complications (e.g. infection, pain, and bleeding). It can often be associated with sexual pain disorder as well.
Dyspareunia is the most common type of PPFSD. Solana-Arellano et al (2008) reported an incidence of 41.3% for dyspareunia in the 60-180 days period after giving birth.1 Postpartum dyspareunia may be due to medical (physical) problems such as a mal-healed perineal or vaginal tear, postpartum infection, cystitis, arthritis, or haemorrhoids, which may get worse after delivery. Moreover, dyspareunia might be caused by psychosocial factors like problems in relationship with the partner, work stress, financial crisis, depression, and anxiety. Dyspareunia, in many cases, can occur as a result of a combination of medical and psychosocial factors. Although, vaginismus is recognised as a different identity, it is usually associated with dyspareunia when it happens in the Puerperium. Vaginismus is the involuntary spasm of the pubococcygeal muscles causing difficult and painful penetration. Sexual desire disorders, Isolated postpartum sexual arousal and orgasmic disorder are rarely seen in postnatal clinics as when they occur they tend to be part of other PPFSDs.
Methods:
Risk Factors for PPFSD:To assess the risk factors for PPFSD a literature review was performed using the National Health Library database including all resources ( AMED, BNI, CINAHL, EMBASE, HEALTH BUSINESS ELITE, HMIC, MEDLINE, and PsycINFO). The MESH word/s used was (postpartum sexual dysfunction OR postpartum dyspareunia OR dyspareunia after delivery OR sexual dysfunction after delivery OR sexual problems after delivery). Other different MESH words (using the word sexuality and/or puerperium) were used as well to expand the search possibilities. Only studies discussing the risk factors of PPFSD after vaginal birth were included. Perineal pain as a complication after episiotomy or tears was differentiated from dyspareunia, and studies on perineal pain after delivery were excluded from the review if they did not discuss the effect of the pain on sexual activity. Effect of Mode of Delivery:Searching the Cochrane library databases has shown no review related to the subject. However, Hicks et al (2004) have conducted a systematic review of literature focused on mode of delivery and the most commonly reported sexual health outcomes, which included dyspareunia, resumption of intercourse, and self-reported perception of sexual health/sexual problems.6 In their systematic review they suggested an association between assisted vaginal delivery and some degree of sexual dysfunction but they reported that associations between Caesarean delivery and sexual dysfunction were inconsistent and continued research was necessary to identify modifiable risk factors for sexual problems related to method of delivery.6 Hicks et al have searched PubMed, CINAHL, and Cochrane databases from January 1990 to September 2003, 6 so we have tried to continue the review by looking into the literature database after that date. To assess the effect of mode of delivery on PPFSD (Caesarean section vs. vaginal birth), a literature review was performed using the National Health Library database including all resource ( AMED, BNI, CINAHL, EMBASE, HEALTH BUSINESS ELITE, HMIC, MEDLINE, and PsycINFO) from October 2003 to January 2010. New MESH words were used, related to comparison between different modes of delivery (Caesarean section, vaginal birth, modes of delivery, sexual dysfunction, sexual disorder, dyspareunia). Additional studies from the reference lists were obtained. Only studies directly compared between caesarean section and vaginal birth in term of assessing the PPFSD were included. Results: Risk Factors for PPFSD:Nineteen studies and one systematic review were retrieved in the period from 01/01/1984 to 01/01/2010. The Cochrane library database review did not have related articles. It is worth mentioning, however, that there was a Cochrane review on postpartum perineal short term pain, not related to sexual activity. Therefore, it was excluded from this review. The systematic review included in this list of literature studies is the Langer and Minetti review on the complications of episiotomy.7 Having systematically reviewed four hundred seventy two articles on the Medline database, they concluded that episiotomy, whether medial or mediolateral, appeared to be the cause of more dyspareunia in comparison to spontaneous perinea tears.7 However, there was no significant difference in the incidence of dyspareunia beyond the three month period after delivery.7 After the latter review, Solana-Arellano (2008) have showed that complications of episiotomy are an important risk factor for postpartum dyspareunia. 1 They have found that infection, dehiscence, and constricted introitus complicated an episiotomy can cause long-term postpartum dyspareunia.1 Moreover, Ejegard et al have investigated the long term quality of women's sex life (12-18 months after first episiotomy-assisted delivery).8 They have reported an adverse effect of episiotomy on women's sex life during the second year post partum .8 Effect of Mode of Delivery:Only eight studies fulfilled the criteria. Full papers were retrieved. There was one randomised controlled trial, one prospective cohort study, one cross-sectional study, and the other 5 were performed retrospectively (4 questionnaire surveys, and 1 interview survey). The total pool sample of patients studied included 3476 cases (1185 Caesarean sections vs. 2291 vaginal deliveries). Four studies aimed to compare PPFSD aspects within other variants, such as pelvic floor morbidity, urinary incontinence, and faecal or flatus incontinence.9, 10, 11, 12 the other four studies purely compared PPFSD variants such as dyspareunia with no other pelvic floor morbidity variants.13, 14, 15, 16 There has been an agreement between the studies on the less sexual problems after caesarean section compared to vaginal delivery in the short term after delivery (i.e. up to 3 months postpartum). However, in long term (i.e. more than 12 months postpartum), the outcome was controversial. A meta-analysis was conducted to compare and summarise the long term PPFSD results ( graph 1) . Graph 1: Forest plot of comparison between studies. Studies to left of the midline were in favour of less long term PPFSD symptoms with caesarean section compared to vaginal delivery. Discussion: From the previous results, birth tract trauma is a risk factor which may lead to PPFSD. Therefore it is a logic presumption to think that avoiding pelvic floor injury by performing a caesarean section especially as an elective mode of delivery may alleviate PPFSD. This presumption, if true, will have very significant clinical and financial implications in practice especially with a pre-existing problem of increasing caesarean section rate in many parts of the developed world. So what research evidence in the literature is available to support or overrule this presumption?. The answer to this question becomes more challenging if we know that the British National Sentinel Caesarean Section Audit showed that 50 percent of consultant obstetricians agreed with the statement ‘‘elective caesarean section will least affect the mother’s future sexual function’’ .17 From the previous meta-analysis, there is little evidence to support that a caesarean section may alleviate long term PPFSD compared to vaginal delivery (p=0,02). But, if we examine the studies’ subgroups and primary/secondary results in more details, this evidence sounds insufficient. Griffiths et al (2006) in their questionnaire survey of a 208 women from the Cardiff Birth Survey Database have showed a significant increase in the prevalence of dyspareunia two years after vaginal birth compared to caesarean section. 9 However, their comparison was between vaginal birth and elective caesarean section as they excluded emergency cases.9 Moreover; they found similar increase in the prevalence of urinary incontinence, incontinence of flatus and subjective depression in the vaginal birth group, which lead us to think whether the dyspareunia was related to these factors and not related to vaginal birth itself. In their paper they did not mention if vaginal birth with no tears or complications was associated with a higher incidence of dyspareunia. In contrast, Klein et al (2005) concluded that women who had intact perineum after vaginal birth had less dyspareunia than those underwent caesarean section.12 However, the incidence of dyspareunia in the latter study was higher among women who had an episiotomy with or without forceps.12 Similar findings were revealed by Buhling et al (2006) and Safarinejad et al (2009), who showed that persistence of dyspareunia longer than 6 months after delivery was the highest after operative vaginal delivery.15, 16 Buhling et al concluded that the incidence of persistent dyspareunia was similar in the caesarean section and the spontaneous vaginal birth without injury groups (approximately 3.5%), whereas, Safarinejad et al (2009) have shown that women after elective Caesarean section had the highest Female Sexual Function Index (FSFI) compared to other groups of delivery including the normal vaginal delivery without injury or episiotomy.15, 16 Although Safarinejad et al (2009) study was robust in many aspects, such as using FSFI and studying the sexual function score for both the women and their partners, I think the main weakness in the study that they included only primiparous women. 16 Therefore, we cannot generalise their findings on women in their second or more pregnancies. Moreover, as a previous caesarean section will increase the operative risk of the successive caesarean sections or will add more risk to the trial of labour if this is opted for in the future, we can expect a higher increased of sexual disorders in the following pregnancies. From previous discussion we found insufficient evidence to advocate a decision of performing a caesarean section on basis of alleviating PPFSD. This evidence is outweighed by the higher risk of caesarean section including bleeding, infection, anaesthesia risk, deep vein thrombosis, pulmonary embolism, impairment of future fertility, risk of scar dehiscence in next labour, injury to bladder and bowels and risk of fetal laceration. Author’s Conclusion: Risk Factors for PPFSD:In this review, there is good evidence to suggest that episiotomy is an important risk factor for short term PPFSD. However, there is little evidence to support a possible long term effect especially if other complications to episiotomy occurred later. Breastfeeding, and the use of progestogen-only pill as contraceptive are other risk factors identified by other studies .18, 19, 20 This may be caused by the low oestrogen level and the consequent dry vagina. 18, 19, 20 Other risk factors for PPFSD include the lack of postpartum sexual health counselling and treatment. 2, 21 Effect of Mode of Delivery:Postpartum female sexual disorder is a common problem which can be overlooked in practice sometimes. Awareness of the problem makes half of the solution. The other half consists of identifying the risk factors, careful antenatal and postnatal counselling and sexual health assessment, and educating women, their partners, and staff about diagnosis and management of the problem. Episiotomy and severe obstetric traumas are the main risk factors. Restricted use of episiotomy and early management of episiotomy complications can play an important role in preventing persistent PPFSD. There is insufficient evidence to suggest caesarean section as a better mode of delivery in term of preventing or alleviating PPFSD.
Headaches associated with or occurring around sexual activity have been recognized since the time of Hippocrates [1, 2]. Wolff [3] discussed headache during sexual activity in 1963. However, these headaches started to be formally reported in the 1970s, first by Kitz in 1970 [4] and then Paulson [5] and Martin [6] in 1974. The first published study was by Lance in 1976 [7].
Classification
This type of headache has been given many different names: benign sex headache (BSH), benign coital headache, coital cephalgia, orgasmic cephalgia, primary headache associated with sexual activity (PHSA), coital ‘thunderclap’ headache, primary thunderclap headache (PTH), orgasmic headache (OH) and preorgasmic headache.
In 2004the International Headache Society [8] classified HSA as a distinct form of primary headache.
These benign HSA are bilateral headaches, precipitated by sexual excitement (masturbation or coitus) occurring in the absence of any intracranial disorder and which can be prevented or eased by ceasing activity before orgasm. Type 1 consists of a bilateral, usually occipital, pressure-like headache that gradually increases with mounting sexual excitement. Type 2 headaches have an explosive, throbbing quality and appear just before or at the moment of orgasm. These often start occipitally but may generalize rapidly [9].
However, there are individuals who experience patterns of HSA that do not fall within the classifications and are included as a subgroup of HSA with unusual psychopathology [10]. For example, Paulson and Klawans [5] described a rare type postural sexual headache after coitus, which is present on standing, eased by lying, accompanied by a low CSF pressure, and persists for several weeks.
International Headache Society diagnostic criteria - ICHD-2(7) classification for HSA
4.4 Primary headache associated with sexual activity
4.4.1 Pre-orgasmic headache
A. Dull ache in the head and neck associated with awareness of neck and/or jaw muscle contraction
and fulfilling criterion B.
B. Occurs during sexual activity and increases with sexual excitement
C. Not attributed to another disorder
4.4.2 Orgasmic headache
A. Sudden severe (“explosive”) headache fulfilling criteria B
B. Occurs at orgasm
C. Not attributed to another disorder
7 Secondary headache disorder
7.2.3 Headache attributed to spontaneous (or idiopathic) low CSF pressure
Prevalence
HSA are not common but it is generally felt that they are under-reported due to patient embarrassment [1] at telling health professionals when their headaches occur. Prevalence in the general population is reported at around 1% [11, 12] and is greater in men than in women, by 3-4 times [11, 13-16]. There appear to be two peak times of onset: in the early 20s and then around age 40 [17]. About 22% of HSA are Type 1 and 78% are Type 2 [18]. The male:female ratio is the same for Type 1 and Type 2 headache.
Pathophysiology
HSA are not clearly understood but by definition lack serious underlying disease. They are however, unpleasant, frightening, repetitive and episodic. The clinical characteristics of Type 1 suggest a relationship with tension/muscular contraction headaches [2, 13, 15, 16]. There is a significant association between the risk of having more than one cluster of HSA and the presence of tension headaches or migraine [11, 14-17, 19-21]. Biehl [11] concluded that the association between migraine and HSA is bilateral. The prevalence of migraine in HSA patients is 25-47% [15, 16, 20]. Ostergaard [14] showed that the presence of concomitant migraine or tension headache was significantly associated with the recurrence of periods lasting weeks to months in which HSA occurred. Patients without another primary headache often have only one HSA period or episode and a more favourable prognosis . Migraine is co-morbid in 30% of Type 2 as opposed to 9% with Type 1. Co-morbidity is also seen in exertional headaches, 35% of Type 2 and 9% Type 1[17, 18]. There can be simultaneous onset of benign exertional headache (BEH) and HSA [22] as well as HSA after a history of BEH [16, 22].
Several drugs have been linked in case reports to sexual headaches associated with neurologic symptoms: Amiodarone [23], birth control pills [24], pseudoephedrine [7] and cannabis [25].An interesting more recent addition to HSA is that resulting from the use of PDE5 medication to assist in erectile difficulties [26, 27].
In type 2 headaches, increased intracranial pressure secondary to a Valsalva maneuver during orgasm has been proposed as a possible mechanism. Blood pressure may increase by 40-100mmHg systolic and 20-50mmHg diastolic during orgasm [7, 28-30]. A possible disruption of autoregulation of the cerebral vasculature has also been proposed [31-33].
Classic presentation
A male patient, middle-aged, in poor physical shape, mildly to moderately overweight, and mildly to moderately hypertensive [34]. In women muscle contraction and psychological factors are often involved [34].
The typical story is that the headache occurs during sexual activity, is bilateral and stops or is less severe if sexual activity stops prior to orgasm. The duration varies from 5 minutes to 2 hours if sexual activity stops and from 3 minutes to 4 hours, with the possibility of milder symptoms up to 48hours, if activity continues.
Differential diagnosis
With the first episode it is absolutely mandatory to exclude potentially life threatening and disabling causes. A thorough history and neurological examination with the option of imaging studies and CSF examination must be conducted.
Type 2 explosive “thunderclap” headaches can be secondary to subarachnoid haemorrhage, aneurysms without obvious rupture, intracerebral haemorrhage, pituitary apoplexy, venous sinus thrombosis, cervical artery dissection, subdural haematoma, haemorrhage into an intracranial neoplasm [35], cerebral tumour [36], intracranial hypotension and hypertension, significant cervical spine disease, and ischaemic stroke [37-43] and these serious conditions need to be excluded before an HSA diagnosis can be given. HSA may present similarly to paroxysmal headaches caused by phaeochromocytoma [44].
Sexual intercourse is reported as a precipitating cause of subarachnoid haemorrhage in 3.8% to 12% of patients with bleeding from a ruptured aneurysm [35].
Course of the disease
The unpredictable clinical course falls into 2 temporal patterns: an episodic course with remitting bouts, and a chronic course [20]. In most cases the headaches occur in bouts that recur over periods of weeks to months before resolving [16, 45].
The episodic type is defined as a bout of at least 2 attacks occurring in ≥ 50% of sexual activity followed by no attack for ≥ 4 weeks despite continuing sexual activity. The chronic course is defined as ongoing HSA attacks for ≥ 12 months without remission of ≥ 4 weeks [20].
Further uncertainty is experienced by the patient as HSA does not necessarily occur in every sexual encounter [7, 19]. A characteristic of HSA is the sporadic vulnerability of patients to the headache. Episodes can occur singly, in clusters or at irregular intervals. Recurrence can occur years later.
The acute HSA attacks are usually short lasting but the overall duration of pain can vary widely [17]. The mean duration of severe pain in HSA is similar (30 minutes) in type 1 and type 2 but the mean duration of milder pain is more prolonged with type 2 (4 hours vs 1 hour). About 15% of patients suffer from severe pain for >4hours needing acute treatment. Severe pain continuing for 2-24 hours occurs in up to 25% of patients with HSA [17]. Patients with episodic HSA compared to chronic HSA have an earlier age at onset and tend to suffer more often from concomitant BEH [20].
About 30% of patients report headaches with masturbation as well as intercourse. There are also reports of HSA occurring exclusively during masturbation [46, 47] and a case of this occurring with nocturnal emission [21].
Overall HSA occurs more commonly when the patient is tired, under stress or attempting intercourse for the second or third time in close succession [48]. HSA appears in bouts lasting weeks to months and can disappear without specific treatment [14, 16]. The number of attacks within one bout ranges from 2 to 50 [17]. About 25% of patients suffer attacks without longer remissions.
Prognosis
Prognosis is usually good for HSA as it is a benign self- limiting disorder and disappears without any specific treatment in the majority of patients [17]. It is usually better if there has been only one attack, especially if it was not associated with any other type of headache.
Frese [20] concluded that episodic HSA occurs in approximately 75% and chronic HSA in approximately 25% of patients. However even in chronic HSA, the prognosis is favourable, with remission rates of 69% in patients followed over 3 years.
Management
A thorough history and examination is mandatory in a first attack.
Referral is warranted if:
Atypical story and suspicious examination
First episode of severe headache where headache still present
A recurrent episode of severe headache with longer than average duration
Neck stiffness, photophobia or vomiting
Altered consciousness or confusion
Focal neurological signs
Previous history of AV malformation, neoplasms or neurosurgery
Investigations
Computed tomography
MRI
Lumbar puncture
Cerebral angiography
Urinary catecholamine
Medical treatment
Turner [49] has provided a good review.
Pre-emptive treatment
Propanolol hydrochloride ( Inderal) is effective in the prophylaxis of HSA[19]. Naratriptan 2.5mg has been reported as useful prior to sexual activity [50] but due to lower absorption rates needs to be taken more than 60 minutes before sexual activity [30]. Indomethacin 25-100mg can be taken 30-60 minutes prior to sexual activity [15, 16, 45, 51] and for acute severe pain management [20] but can cause serious gastrointestinal side-effects and is not tolerated by about 10% of headache patients [52].
Acute treatment
Triptans shorten the attack in about 50% of patients[30]. There is an 80% response rate [30]. Analgesics (ibuprofen, diclofenac, paracetamol, acetylsalicylic acid) given after onset of headache are of limited or no value in nearly all patients [45].
Other triptans, ergots and benzodiazepines have also been reported to have efficacy [5, 24, 53, 54] for acute and pre-emptive treatment for those patients not tolerating indomethacin. Taken 30 minutes before sexual activity they shorten orgasmic headache attacks in 66% of users [30].
Long term prophylaxis for longer lasting bouts or continued attacks
Options include indomethacin 25mg three times a day, propanolol 120-240mg per day, metoprolol 100-200 mg per day and diltiazem 180 mg per day [15, 19, 20, 22, 24, 45]. There is about an 80% response rate [30].
Sexual management
Trauma due to pain associated with sexual activity has the potential to affect immediate and long term satisfaction with sexual activity unless specifically addressed. HSA can be very distressing for both patient and partner with the development of fears around sexual activity and orgasm. Patients may develop patterns of impaired sexual arousal. If these fears are not exposed and dealt with, sexual problems may occur. Secondary avoidance behaviours may become established in the relationship leading to a decrease in couple’s physical affection, eroticism and sexual activity. Patients must be given the opportunity to talk about sexual fears in an ongoing way, especially if HSA is chronic.
The social and relationship history will disclose areas of stress which should be evaluated and managed as best possible. In type 1 HSA where neck and jaw tension may be a factor, conscious relaxation of these muscles during intercourse may help [7]. Relaxation exercises especially concentrating on neck and shoulder tension can be done regularly and particularly before anticipating sexual activity.
Individuals often sense early in the lovemaking process whether or not HSA will occur and encouragement not to pursue orgasm on that occasion can be helpful. Some patients can terminate the headache by stopping the sexual activity or suppressing orgasm and about 51% can lessen the intensity of pain by being more sexually passive [18].
Advice on continuing to engage with the partner despite ceasing or modifying one’s own sexual arousal needs to be given. Having a disappointed or resentful partner increases the distress of the condition so partner needs have to be discussed. Patients often have difficulty talking about sexual issues with both their partner and their doctor, therefore the doctor needs to be the one to raise the subject.
A brief sexual history will outline the love-making practice and modification to sexual positions, especially where neck tension is exaggerated, may help. In one report, the advice to engage in intercourse more frequently but less strenuously resulted in a reduction in headaches [5].
Avoiding sexual activity and strenuous activities until totally symptom free has been recommended by some [13, 22, 24, 55]. This may be difficult to follow as the capricious nature of HSA makes knowing when they have stopped difficult.
Conclusion
HSA are benign, but because they can mimic serious conditions, patients need to be properly assessed before reassurance is given and management of HSA started. Because pain can alter sexual experience and behaviour around sexuality for the patient and the couple, this aspect of patient wellbeing must be addressed by the treating physician for good holistic management. As not everyone is comfortable with addressing sexuality with patients, respectful acknowledgement of the situation and appropriate referral can be a useful approach.
Definition of restraint: a device or medication that is used to restrict a patient’s voluntary movement. Prevalence of physical restraints: up to 17% in acute care settings. Prevalence of chemical restraints: up to 34% psychotropic drug use in long term care facilities. Complications of restraints: include documented falls, decubitus ulcers, fractures, and death. Regulations: require documentation of indications plus failure of alternatives by a licensed professional. Prevention of removal of life sustaining treatment: is a relatively clear indication for restraints. Informed consent: including consideration of risks, benefits, and alternatives is necessary in all cases. Barrier to reducing restraints: a misguided belief that, by use, one is preventing patient injury. Steps can be taken to limit their use: including an analysis of behaviours precipitating their use.
Case study
A 79 year old female nursing home resident with frontotemporal dementia and spinal stenosis has a chronic indwelling catheter for cauda equina syndrome and neurogenic bladder. Attempts to remove the catheter and begin straight catheterization every shift were met by the patient becoming combative with the staff. Replacing the catheter led to repeated episodes of the patient pulling out the catheter. The patient lacks decision making capacity to weigh the risks, benefits, and alternatives; but she clearly doesn’t like having a catheter in. The attending physician instituted wrist restraints pending a team meeting. Unfortunately, attempts by the patient to get free led to dislocation of both shoulders and discharge to the hospital.
Introduction
A restraint is any device or medication used to restrict a patient’s movement. In the intensive care unit, for example, soft wrist restraints may be used to prevent a patient from removing a precisely placed endotracheal tube. A lap belt intended to prevent an individual from falling from a wheelchair in a nursing home is a restraint if the patient is unable to readily undo the latch.1 In the case study above of a catheterized, demented patient, if medication is used to prevent the patient from striking out at staff when performing or maintaining catheterization, then the medication is considered a restraint.
There is little data on efficacy and benefits of restraints1. Even when the indication to use a restraint is relatively clear, the outcome is often opposite of the intention. Consider that restraints used for keeping patients from pulling out their endotracheal tubes are themselves associated with unplanned self- extubation2. Complications of restraints can be serious including death resulting from medications or devices3,4.Use of restraints should be reserved for documented indications, should be time limited, and there should be frequent re-evaluation of their indications, effectiveness, and side effects in each patient. Lack of a Food and Drug Administration (FDA) approved indication for use of medications as restraints in agitated, aggressive, demented patients has led to recommendations that medications in these situations be used only after informed consent with proxy decision makers5. Medical, environmental, and patient specific factors can be root causes of potentially injurious behavior to self or others as in the case study above. To ensure consideration and possible amelioration of these underlying causes, the Center for Medicare and Medicaid Services (CMS ) in 2006 required face to face medical and behavioral evaluation of a patient within one hour after restraints are instituted by a physician (licensed independent practitioner). As a result of controversy surrounding this rule, clarification of that rule in 2007 allowed for a registered nurse or physician assistant to perform the evaluation provided that the physician is notified as soon as possible6 . In depth situational analysis of the circumstances surrounding the use of restraints in individual cases as well as education of the patient, family, and caregivers may lead to the use of less restrictive alternatives7.
Frequency of restraint use
Frequency of restraint use depends on the setting, the type of restraint, and the country where restraint use is being studied. In the acute care hospital setting, reported physical restraint use was 7.4% to 17%.a decade ago8.Two decades ago, in long term care facilities prevalence was reported as 28%-37%.9 . There has been a steady decline over the past several decades coincident with regulation such that, according to the Department of Health and Human Services, it is down to about 5% since newer CMS rules went into effect in 2007. In contrast, some European nursing homes still report physical restraint use from 26% to 56%10,11.
Chemical restraint is slightly more prevalent than physical restraint with a prevalence of up to 34% in long term care facilities in the US prior to regulations12.There is some indication that prevalence may be decreasing, some say markedly, perhaps as a result of government regulation13,12 .Interestingly, one case-control study of more than 71,000 nursing home patients in four states showed that patients in Alzheimer special care units were no less likely to be physically restrained compared to traditional units. Furthermore, they were more likely to receive psychotropic medication14.
Complications of restraint use
The use of chemical and physical restraints is associated with an increase in confusion, falls, decubitus ulcers, and length of stay15,16. Increase in ADL dependence, walking dependence, and reduced cognitive function from baseline has also been reported17. Use of restraints often has an effect opposite the intended purpose of protecting the patient, especially when the intent is prevention of falls18. Physical restraints have even caused patient deaths. These deaths are typically due to asphyxia when a patient, attempting to become free of the restraint, becomes caught in a position that restricts breathing4,19.
Antipsychotic medications may be used as restraints in elderly patients with delirium or dementia who become combative and endanger themselves and others; however, there is no FDA approval for these drugs for this use5. In a meta-analysis, an increased relative risk of mortality of 1.6 to 1.7 in the elderly prompted the FDA to mandate a “black box” label on atypical antipsychotic medications stating that they are not approved for use in the behavioral manifestations of dementia20. Other research suggests that conventional antipsychotics are just as likely to cause death, if not more so3. Forensic research also links antipsychotic medication and patient deaths21. The reported relative risk of falls from these drugs is 1.722. Given the risks, if antipsychotic medications are used at all, they need to be prescribed as part of a documented informed-consent process. Education of patients, families of patients, and facility staff about the harms of restraints is a good first step in a plan to avoid or eliminate their use. Over the past several decades, regulations have arisen in the United States because of complications of restraints and a lack of clear evidence supporting their use.
The regulatory environment in the United States
The Omnibus Budget Reconciliation Act of 1987 (OBRA 87) resulted in regulations that specify the resident’s right to be free of the use of restraints in nursing homes when used for the purpose of discipline or convenience and when not required to treat the resident’s medical symptoms23,24. OBRA87 related regulations also specified that uncooperativeness, restlessness, wandering, or unsociability were not sufficient reasons to justify the use of antipsychotic medications. If delirium or dementia with psychotic features were to be used as indications, then the nature and frequency of the behavior that endangered the resident themselves, endangered others, or interfered with the staff’s ability to provide care would need to be clearly documented24. Comprehensive nursing assessment of problem behaviors, a physician order before or immediately after instituting a restraint, and documentation of the failure of alternatives to restraint are required before the use of a restraint is permitted. The restraint must be used for a specific purpose and for a specified time, after which reevaluation is necessary.
The Joint Commission on Accreditation of Healthcare Organizations (JCAHO) instituted similar guidelines that apply to any hospital or rehabilitation facility location where a restraint is used for physical restriction for behavioral reasons25. In response to the 1999 Institute of Medicine report, To Err is Human, JCAHO focused on improving reporting of sentinel events to increase awareness of serious medical errors. Not all sentinel events are medical errors, but they imply risk for errors as noted in the revised 2007 JCAHO sentinel event definition: A sentinel event is an unexpected occurrence involving death or serious physical or psychological injury, or the risk thereof6. The JCAHO recommends risk reduction strategies that include eliminating the use of inappropriate or unsafe restraints. The recommendations for restraint reduction are prioritized along with items like eliminating wrong site surgery, reducing post-operative complications, and reducing the risk of intravenous infusion pump errors6. It is clear that JCAHO considers placing restraints as a sentinel event to be monitored and reported. CMS and JCAHO have worked to align hospital and nursing home quality assurance efforts especially with respect to the standard concerning face to face evaluation of a patient within one hour of the institution of restraints. They held ongoing discussions that resulted in revised standards for the use of restraints in 200926. Among the agreed upon standards are: policies and procedures for safe techniques for restraint, face to face evaluation by a physician or other authorized licensed independent practitioner within one hour of the institution of the restraint, written modification of the patient’s care plan, no standing orders or prn use of restraints, use of restraints only when less restrictive interventions are ineffective, use of the least restrictive restraint that protects the safety of the patient, renewal of the order for a time period not to exceed four hours for an adult, restraint free periods, physician or licensed independent practitioner daily evaluation of the patient before re-ordering restraint, continuous monitoring, and documentation of strategies to identify environmental or patient specific triggers of the target behavior. The one hour face to face evaluation may be accomplished by a registered nurse provided that the attending physician is notified as soon as possible26.
Indications for use of restraints
The risk of using a restraint must be weighed against the risk of not using one when physical restriction of activity is necessary to continue life-sustaining treatments such as mechanical ventilation, artificial feeding, or fluid resuscitation. Every attempt should be made to allow earlier weaning from these treatments, thereby rendering the restraint unnecessary. Even in cases where the indication is relatively clear, the risks, benefits, and alternatives must be weighed (see Figure).
In an emergency, when it is necessary to get a licensed provider’s order for a restraint to prevent a patient from disrupting lifesaving therapy or to keep a patient from injuring others, an analysis of what may be precipitating the episode is essential. Are environmental factors such as noise or lighting triggering the behavio? Are patient factors such as pain, constipation, dysuria, or poor vision or hearing triggering the disruptive behavior? Is there an acute medical illness? Is polypharmacy contributing? Psychotropic drugs and drugs with anticholinergic activity are common culprits. Patient, staff, family, and other health care providers need to be queried.
One must guard against perceiving the continued need for life-sustaining treatment and the use of restraints as being independent factors, because that misconception can lead to a vicious cycle. For example, a patient who has persistent delirium from polypharmacy and needs artificial nutrition and hydration which perpetuates the need for continued chemical and physical restraints. Correcting the polypharmacy and the restraint as a potential cause of the delirium can break the cycle. When restraints are indicated, one must use the least-restrictive restraint to accomplish what is needed for the shortest period of time. Restraint-free periods and periodic reassessments are absolutely required.
A weaker indication is the use of restraints to prevent patient self-injury when the danger is not imminent. Such an indication exists when a patient repeatedly attempts unsafe ambulation without assistance or when he or she cannot safely ambulate early in the process of rehabilitation from deconditioning or after surgery. In these cases, weighing the risks and benefits of the restraint is more difficult than when considering restraints to maintain life-sustaining treatment.
Even more difficult to justify is the use of restraints to restrict movement to provide nonurgent care. An example might be a patient who repeatedly removes an occlusive dressing for an early decubitus ulcer. In these cases, it is more fruitful to use alternatives to restraints. For example, considering alternatives to a urinary catheter is more important than documenting that restraints are indicated to keep the patient from pulling it out.
If used, the specific indication, time limit, and plan for ongoing reevaluation of the restraint must be clearly documented. Effectiveness and adverse effects must be monitored. Restraint-free periods are also mandatory. The same is true for chemical restraints. Periodic trials of dosage reduction and outcome are mandatory.
Barriers to reducing the use of restraints
Perceived barriers to reducing restraints can be thought of as opportunities to build relationships between patients, physicians, staff, patients’ families, and facility leaders. A legitimate fear of patient injury, especially when the patient is unable to make his or her own decisions, is usually the root motivation to use restraints. Ignorance about the dangers of restraint use results in a sincere, but misguided, belief that one is acting in the patient’s best interest27. Attempts to educate physicians, patients, and staff may not have been made. These barriers are opportunities for the community to work together in creative partnerships to solve these problems. Even in communities where there are no educational institutions, there are opportunities for educational leadership among physician, nursing, and other staff. Conversely, lack of commitment to reducing restraints by institutional leaders will tend to reinforce the preexisting barriers. Regulatory intervention has been a key part of gaining the commitment of institutional leadership when other opportunities were not seized. On the other hand, competing regulatory priorities such as viewing a serious fall injury as a ‘never event’ and simultaneously viewing institution of a restraint as a sentinel event may lead to reduced mobility of the patient18. An example of this would be the use of a lap belt with a patient-triggered release. The patient may technically be able to release the belt, but the restricted mobility may lead to deconditioning and an even higher fall risk when the patient leaves the hospital. In the process of preventing the serious fall injury or ‘never event’ there is, even at the regulatory level, intervention that may not be in the patient’s best interest. These good intentions are, again, a barrier to the reduction of the use of restraints and an opportunity for physician leadership in systems based care collaboration. Physician leadership probably needs to extend beyond educational efforts. Evidence suggests education may be necessary but not sufficient to reduce the use of restraints10.
Reducing the use of restraints
Steps can be taken to reduce the use of restraints before the need for them arises, when the need for restraints finally does arise, and while their use is ongoing.
Programs to prevent delirium, falls in high-risk patients, and polypharmacy are all examples of interventions that may prevent the need for restraints in the first place. Attention to adequate pain control, bowel function, bladder function, sleep, noise reduction, and lighting may all contribute to a restraint-free facility.
When a restraint is deemed necessary, a sentinel event has occurred. Attempts to troubleshoot the precipitating factors must follow. Acute illness such as infection, cardiac, or respiratory illness must be considered when a patient begins to demonstrate falls or begins to remove life-sustaining equipment. Highly individualized assessment of the patient often requires input from physical therapy, occupational therapy, social work, nursing, pharmacy, and family. If root causes are determined and corrected, the need for restraints can be ameliorated and alternatives can be instituted.
The least restrictive alternative should be implemented when needed. For example, a lowered bed height with padding on the floor can be used for a patient who is at risk for falls out of bed in contrast to the use of bedrails for that purpose. Another example is the use of a lap belt with a Velcro release as opposed to a vest restraint without a release. A third example is the use of a deck of cards or a lump of modeling clay to keep the patient involved in an alternative activity to the target behavior that may be endangering the patient or staff. Alternatives to the use of restraints need to be considered both when restraint use is initiated and during their use. Judicious use of sitters has been shown to reduce falls and the use of restraints28. When danger to self or others from patient behaviors and restraints are deemed necessary, a tiered approach has been recommended by Antonelli29 beginning with markers and paper or a deck of cards for distraction and then proceeding up to hand mitts, lap belts, or chair alarms if needed. Vest or limb restraints are the default only when other methods have been ineffective29.
Literature from the mental health field provides some guidance to those attempting to use the least intrusive interventions for older patient behaviors that endanger themselves or others. A combination of system-wide intervention, plus targeted training in crisis management to reduce the use of restraints has been demonstrated to be effective in multiple studies30. In a recent randomized controlled study, one explanation the author gives for the ineffectiveness the educational intervention is that the intervention was “at the ward level unlike other restraint reduction programs involving entire organizations.”10. Research and clinical care in restraint reduction will likely need to be both patient-centered and systems-based in the future.
Case study revisited
Our 79 year old female with frontotemporal dementia and spinal stenosis noted in the above case pulls out her urinary catheter. The physician is called and determines that the patient’s urine has been clear prior to the episode, that she has no fever, nor does she have evidence of acute illness. The patient is likely pulling the catheter out simply because of the discomfort caused by the catheter itself since the patients behavior is at the same baseline as before the catheter was inserted as determined by discussion with the staff. The patient is unable to inhibit her behavior because of the frontotemporal dementia. The physician places a call to the medical power of attorney and explains the risks of bladder infection, bladder discomfort, renal insufficiency, and overflow incontinence from untreated neurogenic bladder. This is weighed against the risk of frequent infections and bladder discomfort from a chronic indwelling urinary catheter, or damage to the urethra from pulling the catheter out. The option of periodic straight catheterization is dismissed by the medical power of attorney as being too traumatic for this demented patient who becomes agitated during this procedure.
The medical power of attorney considers the options and agrees to observation by the staff without the catheter overnight with a team conference the next day. At the conference, it was noted that overnight the patient had several episodes of overflow incontinence in spite being toileted every few hours while awake. The patient had no signs of discomfort and was changed when found to be wet. A bladder scan done at the facility showed a few hundred cubic centimeters of residual urine after the patient was noted wet and changed. The team conference yielded the informed decision to continue checking the patient frequently and changing when wet as well as frequent toileting opportunities.
The patient continued at baseline for twelve weeks until she developed urinary sepsis and the patient’s medical power of attorney was contacted about additional care decisions.
Conclusion
A restraint is any device or medication used to restrict a patient’s movement. Complications of restraints can be serious including death resulting from both medications and devices. Use of restraints should be reserved for documented indications, should be time limited, and there should be frequent re-evaluation of their indications, effectiveness, and side effects in each patient. Analysis of environmental and patient specific root causes of potentially self-injurious behavior can lead to reduction in the use of restraints. Education of the patients, families, and the health care team can increase the use of less restrictive alternatives.
In the first part of this review we considered neurodegenerative and neurobehavioural diseases and the findings that these diseases commonly are associated with systemic and central nervous system bacterial and viral infections.1 In this second part we continue with psychiatric diseases, autoimmune diseases, fatiguing illnesses, and other chronic diseases where chronic infections play an important role.
Psychiatric diseases
Borrelia-associated psychiatric disorders
In addition to neurologic and rheumatologic symptoms Borrelia burgdorferi has been associated with several psychiatric manifestations2, 3 (see also below). Such infections can invade the central nervous system and may cause or mimic psychiatric disorders or cause a co-morbid condition. A broad range of psychiatric conditions have been associated with Lyme disease, including paranoia, dementia, schizophrenia, bipolar disorder, panic attacks, major depression, anorexia nervosa and obsessive-compulsive disorder.4-7 For example, depressive states among patients with late Lyme disease are fairly common, ranging from 26% to 66%.3 It is not known whether B. burgdorferi contributes to overall psychiatric morbidity, but undiagnosed chronic Lyme disease caused by this spirochete is considered a differential diagnosis in patients with certain psychiatric symptoms such as depressive symptoms, lack of concentration and fatigue.
The neuropsychiatric sequelae of chronic Lyme disease remains unclear. Studies were performed, some on large numbers of patients, to investigate whether a correlation exists between chronic Lyme disease (defined by seropositivity) and psychiatric disorders.8-11 Interestingly, different results were reported on the association between B. burgdorferi infection and psychiatric morbidity.8-11 For example, Hájek et al.8 compared the prevalence of antibodies to B. burgdorferi in groups of psychiatric patients and healthy subjects. Among the matched pairs, 33% of the psychiatric patients and 19% of the healthy comparison subjects were seropositive. In contrast, Grabe et al.11 did not find an association between Borrelia seropositivity and mental and physical complaints. In 926 consecutive psychiatric patients that were screened for antibodies and compared with 884 simultaneously recruited healthy subjects, seropositive psychiatric patients were found to be significantly younger than seronegative ones, and this was not found in the healthy controls.10 However, none of the psychiatric diagnostic categories used in this study exhibited a stronger association with seropositivity.10 These findings suggest a potential association between B. burgdorferi infection and psychiatric morbidity, but fail to identify any specific clinical 'signature' of the infection. This might be due to the very low incidence in an endemic region (0.2%, CI 95% 0.0% to 1.1%) as demonstrated in 517 patients hospitalized for psychiatric diseases.9
In addition to serological data, clinical evidence for the association of psychiatric symptoms and post-Lyme disease has also been investigated. If mental and physical complaints in patients were assessed with the von Zerssen's complaint scale using multivariate analyses, the data revealed that definitions of seropositivity were not associated with increased mental or physical complaints.11 In contrast, if the SF-36 was used to determine Quality of Life (QOL) in post-Lyme patients, the average SF-36 physical component summary (40±9, range 29-44) and mental component summary (39±14, range 23-46) of the QOL assessment were worse than the general USA population, and they could be significantly improved by anti-Lyme antibiotics (46% versus 18%, p=0.007).5 Barr et al.12 examined the relation between complaints of memory disturbance and measures of mood and memory functioning in 55 patients with serological evidence of late-stage Lyme borreliosis. There was a significant correlation between subjective memory ratings and self-reported depression (p<0.001) but not with objective memory performance, indicating memory disturbance in chronic Lyme patients. Using a structured psychiatric interview, the Positive and Negative Affect Schedule, the Lyme Symptom Checklist, and a battery of neuropsychological tests in 30 post-Lyme patients, participants did not appear to have an elevated incidence of psychiatric disorders or psychiatric history.13 Their mood, however, was characterized by lowered levels of positive affect and typical levels of negative affect that were similar to affect patterns in individuals with chronic fatigue syndrome (CFS). Similarly, Hasset et al.4, 7 reported on 240 consecutive post-Lyme patients who were screened for clinical psychiatric disorders, such as depression and anxiety. After adjusting for age and sex, these disorders were more common in symptomatic patients than in the comparison group (Odds Ratio=3.54, CI 95% 1.97-6.55, p<0.001), but personality disorders were comparable in both groups.
Although psychiatric co-morbidity and other psychological factors are prominent in post-Lyme patients, it remains uncertain whether these symptoms can be directly attributed to the chronic course of Borrelia infections or to other chronic illness-related factors.
Schizophrenia
Several microbes have been suspected as pathogenetic factors in schizophrenia, such as Chlamydia species, toxoplasma, and various viruses. For example, a number of studies have reported associations between Toxoplasma gondii infection and the risk of schizophrenia with an overall hazard ratio of 1.24.14 In addition, chlamydial infections have been found in 40% of schizophrenic patients compared to 7% in healthy controls.15 These infections represented the highest risk factor yet found to be associated with schizophrenia that was highly significant (Odds Ratio=9.43, p=1.39 x 10-10), especially with Chlamydophila psittaci (Odds Ratio=24.39, p=2.81 x 10-7). Interestingly, schizophrenic carriers of the HLA-A10 genotype were clearly the most often infected with Chlamydia, especially C. psittaci (Odds Ratio=50.00, p=8.03 x 10-5), pointing to a genetically related susceptibility.15 However, skepticism against the role of bacterial infection in schizophrenia was also fostered by the low impact of anti-infectious treatment on the course of disease progression in schizophrenia.16
Genetic backgrounds and viral infections and/or reactivations as well as cytokine-related pathomechanisms have also been proposed as causative for psychiatric disorders, such as schizophrenia. Specific genetic patterns of MICB polymorphism (MHC class I polypeptide-related sequence B, chromosome 6p21) were identified in patients seropositive for CMV and HSV-1.17 Similar polymorphisms were found for the COMT Val158Met related to serological evidence of HSV-1 infections in individuals with bipolar disorder.18 This serologic evidence of HSV-1 infection appeared to be associated with cognitive impairment in individuals with bipolar disorders19 and was found to be an independent predictor of cognitive dysfunction in individuals with schizophrenia.20 In addition, viral exposure during gestation has been described as a risk factor for schizophrenia. Offspring of mothers with serologic evidence of HSV-2 infection were at significantly increased risk for the development of psychoses (Odds Ratio=1.6; CI 95% 1.1-2.3). These results are consistent with a general model of risk resulting from enhanced maternal immune activation during pregnancy.21 However, this was not confirmed in another study.22 Similar contradictory results were observed in a small group of 8 patients with schizophrenia where reactivation of herpesviruses (HSV-1, CMV, EBV, varicella-zoster virus and human HHV-6) and other viruses (measles, rubella, mumps, influenzaA and B and Japanese encephalitis viruses) during acute onset or exacerbation of schizophrenia was investigated, but none of these viruses were detected in these patients.23 Also, a search for HSV-1 or varicella zoster virus infection in postmortem brain tissue from schizophrenic patients did not reveal evidence of persistent CNS infections with these viruses.24
Schizophrenic patients show a number of cytokine changes that may be important in their condition. For example, differences in interleukin-2, -4 and –6, among other cytokines, have been seen in schizophrenic patients.25-27 Often these changes in cytokines or cytokine receptors have been linked to associated genetic changes found in schizophrenia.28-30 Monji et al.31 recently reviewed the evidence for neuroinflammation, increases in pro-inflammatory cytokines and genetic changes in schizophrenia and concluded that these changes are closely linked to activation of microglia. Although the microglia comprise only about 10% of the total brain cells, they respond rapidly to even minor pathological changes in the brain and may contribute to neurodegeneration through the production of pro-inflammatory cytokines and free radicals. CNS infections could also activate microglia and cause similar events.
Neuropsychiatric Movement Disorders
Giles de la Tourette’s syndrome (TS) is a neurological condition that usually begins in childhood and results in involuntary sounds or words (vocal tics) and body movements (movement tics). An association between infection and TS has been repeatedly described.32 Abrupt onset of the disease, usually after infection, was noted in up to 11% of these patients.33-34 A role for streptococcal infections (PANDAS, see below) as causative or mediating agent in TS was established several years ago.35 Additionally, the involvement of other infectious agents, such as B. Burgdorferi or M. pneumoniae, has been described in case reports and small studies. For example, comparing 29 TS patients with 29 controls revealed significantly elevated serological titers in TS patients (59% versus 3%). This higher proportion of increased serum titers, especially IgA titers, suggested a putative role for M. pneumoniae in a subgroup of patients with TS.36 In predisposed persons, infection with various agents including M. pneumoniae should be considered as at least an aggravating factor, but an autoimmune reaction has to be taken into account in TS patients. In addition, co-infections with toxoplasmosis have been described in a few case reports of obsessive-compulsive disorder (OCD).37 As mentioned above, streptococcal infections are likely to play a pivotal role in these syndromes.35
The pathogenic mechanism may be secondary to an activation of the immune system, resulting in an autoimmune response. This will be discussed in the next section.
Autoimmune Diseases
Infections are associated with various autoimmune conditions.38-40 Autoimmunity can occur when infections like cell-wall-deficient bacteria are released from cells containing parts of cell membranes that are then seen as part of a bacterial antigen complex, or bacteria can synthesize mimicry antigens (glycolipids, glycoproteins or polysaccharides) that are similar enough in structure (molecular mimicry) to stimulate autoimmune responses against similar host antigens. Alternatively, viral infections can weaken or kill cells and thus release cellular antigens, which can stimulate autoimmune responses, or they can incorporate molecules like gangliosides into their structures.
In addition to molecular mimicry, autoimmunity involves several other complex relationships within the host, including inflammatory cytokines, Toll-like receptor signalling, stress or shock proteins, nitric oxide and other stress-related free radicals, among other changes that together result in autoimmune disease.38, 39
Guillain-Barré syndrome
Guillain-Barré syndrome (GB) is a demyelinating autoimmune neuropathy often associated with bacterial infections.40 Symptoms include pain, muscle weakness, numbness or tingling in the arms, legs and face, trouble speaking, chewing and swallowing. Of the types of infections found in GB, Campylobacter jejuni, Mycoplasma pneumoniae and Haemophilus influenzae are often found.39 For example, Taylor et al.41 found serological evidence of C. jejuni in 5 of 7 patients with GB and other motor neuropathies, and Gregson et al.42 found anti-ganglioside GM1 antibodies that cross-reacted with C. jejuni liposaccharide isolates. When infections were examined in GB cases in India, Gorthi et al.43 found that 35% and 50% of GB patients had serological evidence of C. jejuni and M. penumoniae infections, respectively, while one-third of cases showed evidence of both infections. In Japan Mori et al.44 found that 13% of GB patients had antibodies against Haemophilus influenzae . Autoantibodies stimulated by infections found in GB patients can cross-react with nerve cell gangliosides (anti-GM1, anti-GM1b, anti-GD1a, among others), and these are thought to be important in the pathogenesis of GB.45 Indeed, injection of C. jejuni lipo-oligosaccharide into rabbits induces anti-gangliosides and a neuropathy that resembles acute motor axonal neuropathy.46
Viruses have also been found to be associated with GB.40 Examples are: CMV,47 HIV,48 herpes simplex virus,49 West Nile virus,50 and HHV-6.51
Paediatric autoimmune neuropsychiatric disorders associated with Streptococci ('PANDAS')
Streptococcal infections in children are usually benign and self-limited. In a small percentage of children, however, prominent neurologic and/or psychiatric sequelae can occur. Post-streptococcal basal ganglia dysfunction has been reported with various manifestations, all of which fall into a relatively well-defined symptom complex or syndrome called paediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS).52
Evidence from past studies indicates that adults and children with a symptom course consistent with PANDAS experience subtle neuropsychological deficits similar to those of primary psychiatric diagnosis of OCD and TS.53 PANDAS are now considered as a well-defined syndrome in which tics (motor and/or vocal) and/or OCD are consistently exacerbated in temporal correlation to a group A beta-hemolytic streptococcal infection. However, the pathological relationship between OCD or tics/TS in childhood to antecedent group A Streptococci is still not fully understood.52
In an epidemiological investigation Leslie et al.54 assessed whether antecedent streptococcal infection(s) increase the risk of subsequent diagnosis of OCD, TS, other tic disorders, attention-deficit hyperactivity disorder (ADHD) or major depressive disorder (MDD). Children with newly diagnosed OCD, TS, or tic disorder were more likely than controls to have had a diagnosis of streptococcal infection in the previous year (Odds Ratio=1.54, CI 95% 1.29-2.15). Previous streptococcal infection was also associated with incident diagnoses of ADHD (Odds Ratio=1.20, CI 95% 1.06-1.35) and MDD (Odds Ratio=1.63, CI 95% 1.12-2.30).54 Similar results were found in a retrospective, cross-sectional, observational study of 176 children and adolescents with tics, TS, and related problems.55 In a case-control study of children 4 to 13 years old patients with OCD, TS, or tic these disorders were more likely than controls to have had prior streptococcal infection (Odds Ratio=2.22; CI 95% 1.05-4.69) in the 3 months before onset date. The risk was higher among children with multiple streptococcal infections within 12 months (Odds Ratio=3.10; CI 95% 1.77-8.96).56 Having multiple infections with group A beta-hemolytic Streptococcus within a 12-month period was associated with an increased risk for TS (Odds Ratio=13.6; CI 95% 1.93-51.0). Similar results were found in patients with typical symptoms of Tourette's syndrome.57 The frequency of elevated anti-streptolysin O titers was also significantly higher (p=0.04) in patients with attention-deficit hyperactivity disorder (64%) than in a control group (34%).58
Sydenham's chorea is one manifestation of post-streptococcal neuropsychiatric movement disorders. A pathogenic similarity between Sydenham's chorea, TS and other PANDAS has been suggested since some patients can present with one diagnosis and then evolve with other neuropsychiatric conditions.59 These observations support a role of group A streptococcal infection and basal ganglia autoimmunity. Anti-basal ganglia antibodies that are associated with serologic evidence of recent streptococcal infection were found as potential diagnostic markers for this group of disorders, which includes Sydenham's chorea as the prototype.60
However, contradictory results were also reported.61 For example, an association between symptom exacerbations and new group A beta-hemolytic streptococcus infections among 47 paediatric patients with TS and/or OCD was not observed.59 In addition, the failure of immune markers for streptococcal infections to correlate with clinical exacerbations in a small study of children with paediatric autoimmune neuropsychiatric disorders raised concerns about the viability of autoimmunity as a pathophysiological mechanism in these syndromes.62 However, in a second study the same group reported that patients who fit published criteria for paediatric autoimmune neuropsychiatric disorders associated with streptococcal infections represented a subgroup of those with chronic tic disorders and OCD. These patients may be vulnerable to group A beta-hemolytic Streptococcus infection as a precipitant of neuropsychiatric symptom exacerbations. 63
Taken together, these findings provide epidemiologic evidence that some paediatric-onset neuropsychiatric disorders, including OCD, tic disorders, ADHD, and MDD, may be, at least partially, related to prior streptococcal infections. Group A beta-hemolytic Streptococcus infections are likely not the only event associated with symptom exacerbations for PANDAS patients, but they appear to play a role at least in a subgroup of these children. A potential genetic susceptibility for these post-infectious complexes has been recently proposed.64
The recent recognition that these paediatric neurobehavioural syndromes have infectious and/or immunologic triggers has pointed to important new avenues for their management.
Chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) is a fatiguing illness characterised by unexplained, persistent long-term disabling fatigue plus additional signs and symptoms, including neurophysiological symptoms.65 Brain imaging studies have shown that CFS/ME patients are dysfunctional in their ventral anterior cingulate cortex, and they also have other brain MRI abnormalities.66, 67 In addition, CFS/ME patients also have immunological and inflammation abnormalities, such as alternations in natural killer cell function68, 69 and cytokine profiles.70, 71 In addition, the hypothalamo-pituitary-adrenal axis, which plays a major role in stress responses, appears to be altered in CFS/ME.72
Most, if not all, CFS/ME patients have multiple chronic bacterial and viral infections.73-80 For example, when patients were examined for evidence of multiple, systemic bacterial and viral infections, the Odds Ratio for this was found to be 18 (CI 95% 8.5-37.9, p< 0.001).75 In this study CFS/ME patients had a high prevalence of one of four Mycoplasma species (Odds Ratio=13.8, CI 95% 5.8-32.9, p< 0.001) and often showed evidence of co-infections with different Mycoplasma species, C. pneumoniae (Odds Ratio=8.6, CI 95% 1.0-71.1, p< 0.01) and HHV-6 (Odds Ratio=4.5, CI 95% 2.0-10.2, p< 0.001).75 In a separate study the presence of these infections was also related to the number and severity of signs and symptoms in CFS/ME patients, including neurological symptoms.77 Similarly, Vojdani et al.76 found Mycoplasma species in a majority of CFS/ME patients, but this has not been seen in all studies.81 Interestingly, when European CFS/ME patients were examined for various Mycoplasma species, the most common species found was M. hominis,82 whereas in North America the most common species found was M. pneumoniae,75, 77 indicating possible regional differences in the types of infections in CFS/ME patients. In addition to Mycoplasma species, CFS/ME patients are also often infected with B. burgdorferi,80 and as mentioned above, C. pneumoniae.75, 77, 83
Other infections are also found in CFS/ME patients, such as viral infections: CMV,84 parvovirus B19,78 enterovirus79 and HHV-6.75, 77, 85-88 For example, Ablashi et al.88 found that 54% of CFS/ME patients had antibodies against HHV-6 early protein, compared to 8% of controls. Similarly, Patnaik et al.86 found that 77% of CFS/ME patients were positive for HHV-6 early antigen IgG or IgM antibodies, whereas only 12% of control subjects had IgG or IgM antibodies to HHV-6 early antigen. Recently a new retrovirus, XMRV, was found in mononuclear blood cells of 67% of 101 chronic fatigue syndrome patients compared to only 3.7% of healthy controls. Cell culture experiments determined that the patient-derived virus was infectious and could possibly be transmitted.89
Gulf War illnesses
GWI is a syndrome similar to CFS/ME.90 In most GWI patients the variable incubation time, ranging from months to years after presumed exposure, the cyclic nature of the relapsing fevers and the other chronic signs and symptoms, and their subsequent appearance in immediate family members, are consistent with an infectious process.90, 91 GWI patients were exposed to a variety of toxic materials including chemicals, radiochemicals and biologicals so not all patients are likely to have infections as their main clinical problem. Neurological symptoms are common in GWI cases.90 Baumzweiger and Grove92 have described GWI as neuro-immune disorder that involves the central, peripheral and autonomic nervous systems as well as the immune system. They attribute a major source of the illness to brainstem damage and central, peripheral and cranial nerve dysfunction from demyelination. They found GWI patients have muscle spasms, memory and attention deficits, ataxia and increased muscle tone.92
Bacterial infections were a common finding in many GWI patients.90 Mycoplasmal infections were found in about one-half of GWI patients, and more than 80% of these cases were PCR positive for M. fermentans.90, 91, 93-95 In studies of over 1,500 U.S. and British veterans with GWI, approximately 45% of GWI patients have PCR evidence of such infections, compared to 6% in the non-deployed, healthy population. Other infections found in GWI cases at much lower incidence were Y. pestis, Coxiella burnetii and Brucella species.90
When we examined the immediate family members of veterans with GWI who became sick only after the veteran returned to the home, we found that >53% had positive tests for mycoplasmal infections and showed symptoms of CFS/ME. Among the CFS/ME-symptomatic family members, most (>80%) had the same Mycoplasma fermentans infection as the GWI patients compared to the few non-symptomatic family members who had similar infections (Odds Ratio=16.9, CI 95% 6.0-47.6, p<0.001).91 In contrast, in the few non-symptomatic family members that tested Mycoplasma-positive, the Mycoplasma species were often different from the species found in the Gulf War Illness patients (M. fermentans).The most sensible conclusion is that veterans came home with M. fermentans infections and then transmitted these infections to immediate family members.91
Some other infectious diseases with neurological aspects
Lyme Disease
Lyme disease is caused by a tick bite and the entry of the spiral-shaped spirochete B. burgdorferi as well as other co-infections.96 Lyme disease is the most common tick-borne disease in North America. After incubation for a few days to a month, the Borrelia spirochete and co-infections migrate through the subcutaneous tissues into the lymph and blood where they can travel to near and distant host sites, including the central nervous system.3, 97-99 Transplacental transmission of B. burgdorferi and co-infections can occur in pregnant animals, including humans, and blood-borne transmission to humans by blood transfusion is likely but unproven. The tick-borne co-infections associated with Lyme disease can and usually do appear clinically at the same time, complicating clinical dignoses.100
Lyme disease signs and symptoms eventually overlap with the signs and symptoms of other chronic illnesses, and patients are often diagnosed with illnesses like CFS/ME, chronic arthritis or a neurological disease.80,97-100 About one-third of cases with Lyme disease start with the appearance of a round, red, bulls-eye skin rash (erythema migrans) at the site of the tick bite, usually within 3-30 days.100 Within days to weeks mild flu-like symptoms can occur that include shaking chills, intermittent fevers and local lymph node swelling. After this localised phase, which can last weeks to months, the infection can spread to other sites resulting in disseminated disease. In the disseminated (late) phase patients present with malaise, fatigue, fever and chills, headaches, stiff neck, facial nerve palsies (Bell’s palsy) and muscle and joint pain, and other signs and symptoms.100-104
The disseminated (late) phase of Lyme disease is a chronic, persistent disease with ophthalmic, cardiac, musculoskeletal, central nervous system and internal organ invasion. When it involves the central and peripheral nervous systems, it is often termed neuroborreliosis.100, 104 At this late stage, arthritis, neurological impairment with memory and cognitive loss, cardiac problems (such as myocarditis, endocarditis causing palpitations, pain, bradycardia, hypertension) and severe chronic fatigue are usually apparent.80, 100-102 The signs and symptoms of the chronic (late) phase of the disease usually overlap with other chronic conditions, such as CFS/ME, chronic arthritis, as well as neurodegenerative diseases, causing confusion in the diagnosis and treatment of the chronic phase in patients with Lyme Disease.80, 97, 100, 105 Patients with late stage neuroborreliosis exhibit neuropathologic and neuropsychiatric disease similar to some of the neurodegenerative diseases discussed in previous sections.1
Diagnostic laboratory testing for Lyme disease at various clinical stages is not fool-proof, and experts often use a checklist of signs and symptoms and potential exposures, along with multiple laboratory tests to diagnose Lyme disease.104 The laboratory tests include serology, Western blot analysis of B.burgdorferi associated bands, PCR analysis of blood and the nonspecific decrease in CD-57 natural killer cells. Unfortunately, similar to other intracellular bacteria, Borrelia spirochetes are not always released into the blood circulation or other body fluids, making the very sensitive PCR method less than reliable for diagnosing Lyme Borrelia with blood samples. Lebech and Hansen106 found that only 40% of cerebrospinal fluid samples from patients with Lyme neuroborreliosis were positive for B.burgdorferi by PCR.
Co-infections in Lyme disease are important but, in general, have not received the attention that B. burgdorferiattracts. Some of the Lyme Disease co-infections on their own, such as M. fermentans, have been shown to produce signs and symptoms comparable to B. burgdorferi infections.80, 102
The most common co-infections found in Lyme disease are species of Mycoplasma, mostly M. fermentans, present in a majority of cases.80, 103, 107 In some cases multiple mycoplasmal infections are present in patients with Lyme disease,80 while other common co-infections include Ehrlichia species, Bartonella species and Babesia species. Such co-infections are present in 10-40% of cases.103, 104, 108-112Ehrlichia and Bartonella species are usually found along with Mycoplasma species in Lyme disease.94, 98, 108-111 Bartonella species, such as B.henselae,111 which also causes cat-scratch disease,113 are often found in neurological cases of Lyme disease.100, 111
Protozoan co-infections have been found with B.burgdorferi, such as intracellular Babesia species.100, 108, 109, 112, 114 The combination of Borrelia, Mycoplasma and Babesia infections can be lethal in some patients, and ~7% of patients can have disseminated intravascular coagulation, acute respiratory distress syndrome and heart failure.109
Brucellosis
Brucellosis is a nonspecific clinical condition characterized by intracellular Brucella species infection.115 Approximately 40% of patients with Brucella spp. infections have a systemic, multi-organ chronic form of brucellosis that is similar to CFS/ME in its multi-organ signs and symptoms.115, 116Brucella infections can invade the central nervous system and cause neurological symptoms.117
Brucella species cause infections in animals, and often humans get the infections from prolonged contact with infected animals. Thus these bacteria are zoonotic, they are capable of being transmitted from animals to humans. Although there are at least eight species of Brucella that are pathogenic, only B. melitensis, B. abortus, B. suis and B. canis have been reported to be pathogenic in humans.116
When CFS/ME patients were examined for the presence of Brucella spp. infections, approximately 10% showed evidence by PCR of Brucella spp. infections (Odds Ratio=8.2, CI 95% 1-66, p<0.01).118 Interestingly, urban CFS/ME patients with Brucella infections were not as prevalent as rural patients with Brucella infections (Odds Ratio=5.5, CI 95% 3-23.5, p<0.02), while control subjects had very low (1.4%) rates of infection. Co-infections with Mycoplasma species were also found in Brucella-positive CFS/ME patients.118
Final comments to part 2
The progression, and in some cases, the inception of many chronic diseases are probably elicited by various bacterial and viral infections.1, 39, 40, 119 Even if infections are not directly involved in the pathogenesis of these diseases, patients with chronic conditions are at risk of a variety of opportunistic infections that could result in co-morbid conditions or promote disease progression. Infections can complicate diagnosis and treatment, and patients with late-stage disease with complex neurological manifestations, such as meningitis, encephalitis, peripheral neuropathy, psychiatric conditions, or with other signs and symptoms could have infections that are not recognized or treated.
Patients with chronic diseases are particularly difficult to treat using single modality approaches, and this is particularly true for patients who also have multiple chronic infections.103, 109 The multi-focal nature of chronic diseases and the fact that often treatments are given to suppress signs and symptoms, rather than treat causes of the disease or its progression, have resulted in incomplete or ineffective treatments. On the other hand, even if the causes of chronic diseases are known, by the time therapeutic intervention is undertaken, it may be entirely too late to use approaches that should work on the disease if chronic infections were not present. Moreover, if complex, chronic infections are ignored or left untreated, recovery may be difficult, if not impossible to achieve.
At the moment the evidence that particular or specific types of infections are responsible for the inception or pathogenesis of chronic diseases is inconclusive.119 One of the problems that arises in trying to prove this hypothesis is that not all patients appear to have similar chronic infections. Some individuals can harbour chronic infections without any observable signs or symptoms. Although the incidence of chronic infections of the types discussed in this review in symptom-free individuals is generally very low, usually only a few percent,74-76, 120 that does not prove that they are important in pathogenesis. Since patients with chronic diseases have been identified that do not have easily diagnosed chronic infections, most researchers have concluded that infections are not involved in the pathogenesis of chronic diseases. Unfortunately, the tools available to find chronic infections are not optimal, and many patients are likely go undiagnosed with chronic infections for purely technical reasons.1,119-121
In the history of medicine animal models of disease have provided useful information that could not be obtained through clinical studies alone. Indeed, the field of chronic diseases could benefit from the greater use of relevant animal models. We suggest that to be useful, the pathogenesis of the animal models of disease must be similar to the pathogenesis of human disease and the animal models must have a similar response to therapy as humans. Thus such models are only relevant if they closely mimic human disease and its response to treatment. For example, the infection of non-human primates with neuropathologic microorganisms, such as Mycoplasma fermentans, resulted in brain infections and fatal diseases with clinically typical neurological signs and symptoms.122 These primates also respond to therapies that have been used successfully to treat humans.93,123 Thus this particular model may be useful if it can be reproucibly infected with specific microorganisms and later develop neurological signs and symptoms that closely mimic chronic human neurological diseases. Future efforts to determine the relationship between specific infections and the pathogenesis of various chronic diseases may well depend on the further development of relevant animal models.
Malnutrition is defined as state of nutrition in which there is a deficiency or excess of energy, protein and other nutrients causing measurable adverse effects on tissue/body form, function and clinical outcome. (1) It is recognised that 30% of in-hospital patients are malnourished (undernourished) on admission and the majority of these will lose further weight while in hospital. (2) In this review article the term undernutrition will be used instead of the generalised term malnutrition. Undernutrition develops due to increased losses (vomiting, diarrhoea, malabsorption), decreased intake (anorexia, vomiting, nausea, dysphagia), increased requirements (catabolic state) or a combination of all these processes.
Is undernutrition important?
Consequences of undernutrition include reduced muscle mass, impaired immune function, poor tissue viability, poor clinical outcome and psychosocial effects. (3) Reduced muscle mass decreases cardio-pulmonary function, lean muscle mass and muscle weakness. Impaired immune function increases infection and sepsis risk. Poor tissue viability can cause pressure sores and poor wound healing. Undernutrition can amplify the length of hospital stay. Psychosocial effects include altered mood and poor quality of life. (4) It is therefore essential that undernutrition is properly treated to diminish patient morbidity and mortality.
How to recognise undernutrition:
1. Malnutrition screening tools
Screening tools should be performed easily with low-level staff training. Hospital patients at risk of undernutrition are identified by screening methods such as MUST (Malnutrition Universal Screening Tool) (5), SGA (Subjective Global Assessment) (6) or MNA (Mini Nutritional Assessment – validated for > 65 years) (7). These screening methods usually consider current body mass index (BMI), recent weight loss and possible future weight loss. MUST is currently well advertised as a nutritional screening tool within UK hospitals. The British Association of Parenteral and Enteral Nutrition have endorsed this 5 step-screening tool. The MUST score (low, moderate, high risk) has been shown to correlate with mortality (low risk group 8% vs. high risk group 32%, p 0.01) and length of hospital stay (low risk group 15 days vs. high risk group 28 days, p 0.02) (8). See Table 1.
Table 1
MUST Screening tool (5)
Step 1: BMI (can use alternatives such as ulna length for height or mid-upper arm circumference (MUAC) as approximation for BMI: MUAC <23 cm = BMI 20kg/m2, MUAC >32cm = BMI >30 kg/m2)
Step 2: Percentage weight loss
Step 3: Establish Acute Disease Effect and score
Step 4: Add scores step 1,2,3 together to obtain overall risk of malnutrition
Step 5: Develop care plan
2. Assessment of nutritional status:
If a patient is found to be at risk of undernutrition, following screening, then a formal nutritional assessment ensues. This assessment involves anthropometrics, biochemical testing, clinical methods and dietary history.
a) Anthropometrical data: appropriately trained staff can perform weight, height, waist circumference, mid-upper arm circumference and skinfold thickness measurements. Indices can subsequently be calculated. These include percentage weight loss, BMI and waist-hip ratio.
b) Biochemical data: information acquired from blood testing includes renal function as a marker of hydration. Also sepsis markers including CRP, ESR and WBC’s are valuable surrogate markers for stress response. Albumin is a poor marker of nutritional status. (9)
c) Clinical: medical history including past and present is valuable. It is essential to obtain plans regarding fasting for investigations. Knowledge of current treatment that may cause decreased intake or increased losses is essential.
d) Dietary History: there are assorted techniques of obtaining dietary history. Mostly recall, record diary and food frequency questionnaires methods are utilised.
The overall nutritional assessment entails considering all the information obtained from these different methods. Subsequently a clinical decision is reached regarding the overall nutritional status.
How to assess nutritional requirements:
In-patient energy requirements are calculated using a combination of:
a) basal metabolic rate equations such as Schofield, Harris Benedict and Ireton Jones
b) stress factors or weight gain/loss
c) combined factor for activity level and diet induced thermogenesis.
The basal metabolic rate is typically calculated using the Schofield equation (10). Schofield estimates basal metabolic rate of a healthy individual. An adjustment is then made for stress or weight gain/loss. Stress factors have been published for various clinical conditions including brain injury, infection, pancreatitis and surgery. Finally a combined factor (activity and diet-induced thermogenesis) is added to calculate total energy requirements. This combined factor is adjusted depending on patient mobility. Community patient’s energy requirements are calculated using a separate method. Occupational and non-occupational activity is estimated to determine a physical activity level which is multiplied by basal metabolic rate to achieve overall energy requirements.
Nitrogen/protein requirements are estimated using current patient clinical state i.e. hypermetabolic, depleted or normal state. Normal state nitrogen requirements are 0.14-0.20 g/kg/day. Depleted state patients nitrogen requirements are 0.20-0.40 g/kg/day. (1g nitrogen = 6.25g protein)
Methods of treating undernutrition
Following identification of undernutrition a patient’s treatment can be instituted. Undernutrition is typically treated using a graded stepwise approach and subsequently monitoring response. If however it appears clinically obvious that the first steps would not be advantageous then treatment can be commenced at a more aggressive phase. Improving energy intake using the following methods can treat undernutrition:
Step 1. Increase frequency and quantity of food intake. Consider nutrient dense foods. Encourage foods that are energy and nutrient dense such as meat, fish, cheese, eggs, dairy produce and snack foods.
Step 2. Increase nourishing drinks including milk based drinks, soups, fruit juices and sugary drinks. Nourishing drinks are simple to make and can provide high calories in a small quantity.
Step 3. Food fortification. This essentially increases the energy density of foods by adding high-energy components such as addition of cheese, milk powder, cream, jam and butter to other foods.
Step 4. Supplements. These can be milk, yoghurt or juice based. They contain varying calories (1-2kcal/ml) and protein (4-6g protein/100ml). Supplements are very useful at boosting energy intake. Most are nutritionally complete but others contain calories only. Examples include Ensure Plus, Fortisip, Calogen and Calshake.
Step 5. Enteral feeding. Enteral feeding is used to provide either supplementary or complete nutrition to patients that are unable to maintain adequate nutrition by oral route. It is only likely to benefit malnourished patients or those at risk of malnutrition. This includes patients that have had a failed trial of diet modification or supplementary feeds or patients at pulmonary aspiration risk from oral nutrition.
Step 6. Parenteral nutrition. The development of parenteral nutrition in the 1960’s meant that feeding was possible even in patients that did not have a functioning gastrointestinal tract. Although it is mentioned in this article as a final step in nutritional support it may also be appropriate to use early depending on clinical scenario.
Enteral versus Parenteral feeding
Enteral feeding produces gastro-intestinal luminal contents that can decrease the possibility of gut atrophy. Maintaining a normal intestinal mucosa reduces the hazard of bacteria and toxins crossing the gastro-intestinal wall and therefore can decrease proinflammatory mediator levels. Compared with parenteral nutrition, enteral feeding attenuates the acute phase response and improves disease severity in acute pancreatitis. (11) Hernandez et al have shown that enteral feeds decrease gut mucosal atrophy in critically ill patients. (12) A meta-analysis has shown that in acute pancreatitis use of EN was associated with a significant reduction in infectious morbidity, hospital length of stay, and a trend toward reduced organ failure when compared with use of parenteral nutrition. (13) However it is important to note that many of the studies involving parenteral nutrition had full dose daily calorie intake whereas enteral feeding studies were less likely to reach estimated energy requirements. This is significant since ill stressed patients should not be given full calorie energy requirements in the first 24-48 hours of commencing feeding. Therefore the parenteral feeding groups were disadvantaged in that the patients were overfed initially and received excess energy calorie intake. National Institute of Clinical Excellence states that parenteral nutrition is only to be used in patients with “inadequate or unsafe oral and/or enteral nutritional intake and a non-functional, inaccessible or perforated (leaking) gastrointestinal tract.” (14) Enteral nutrition can be provided by a number of methods including nasogastic tubes, nasojejunal tubes, gastrostomy tunes (including PEG tubes) and jejunostomy tubes (including PEG-J and D-PEJ). Parenteral nutrition can be given peripherally for approximately 14 days but central access should be used if greater than 14 days. Refeeding syndrome
Refeeding syndrome is a potentially lethal condition with severe electrolyte and fluid shifts with resulting metabolic disturbances in malnourished patients. (15) It is caused by refeeding in malnourished patients with resulting insulin release and intracellular movement of potassium, phosphate and magnesium and increased thiamine uptake. It is essential that patients are correctly identified (See Table 2) and electrolytes corrected and intravenous Vitamin B and C is given. Feeding should be initiated slowly. Fluid balance and electrolytes should be monitored closely.
Table 2
NICE guidelines: Risk of refeeding syndrome (14)
>1 of
BMI <16.5 kg/m2
Weight loss >15%
No food intake 10 days
Decreased magnesium/phosphate/potassium
>2 of
BMI <18.5 kg/m2
Weight loss <10%
No food intake 5 days
Alcohol abuse, Insulin, Chemotherapy
Cardiovascular disease and nutrition
Research has confirmed link between cardiovascular disease and serum cholesterol. The NHANES II data has shown that patients who died form myocardial infarction had increased cholesterol. (16) Hooper et al confirmed that dietary fat is linked to cardiovascular risk. (17) This group demonstrated that decreasing total dietary fat over 6 month period reduced cardiac events by 16%. The Seven Countries Study illustrated that cardiovascular mortality was linked to saturated fat intake. (18)
There has been recent interest in the role of the Mediterranean diet in the prevention of cardiovascular disease. (See Table 3)
Table 3
Mediterranean Diet Studies
Population Studies CARDIO2000. Panagiotakos et al 2002. (28) Greece. 2000-2002. CVD group versus control group. Intake of Mediterranean diet type foods significantly decreased risk of developing CVD. The daily use of olive oil and consumption of vegetables, legumes, cereals and fish was associated with 23% risk of developing acute coronary syndromes. Trichopoulou. 2005. (29) Studies relationship between Mediterranean diet and survival in 1302 Greek patients with CVD. Patients with higher compliance with Mediterranean diet had lower cardiac mortality. Martinez-Gonzalez. 2002. (30) Case-control study. Examined the relationship between a Mediterranean diet and risk of first MI. Better compliance with Mediterranean diet lowered risk of MI.
Intervention Studies Medi-RIVAGE. Dietary intervention Study. (31) 3 month diet. Compared Mediterranean diet and low fat diet. There was greater reduction in cardiovascular risk in Mediterranean diet group compared to low fat group (15% vs 9%). GISSI-Prevenzione. Barzi et al. (32) Supplement of Vit-E and omega-3 FA’s given to survivors of recent MI. Also informed to increase intake of Mediterranean diet foods. Outcomes showed that lower chance of premature death if higher intake of Mediterranean foods. Lyon Diet Heart Study. (33) Key study in relation to Mediterranean diet and CVD. Secondary prevention trial. Examined the effects of a Mediterranean diet in survivors of first MI. Experimental group supplied with margarine with high levels of alpha-linolenic acid (n-3 FA). Mediterranean diet group had lower total mortality (70%).
Patients were previously educated regarding low fat diet benefits but Mediterranean diet appears to have enhanced beneficial effects. There is however some complexity with definition of a Mediterranean diet. The diet is based on dietary patterns of Greece, Crete and Southern Italy in 1960’s. This diet was abundant in plant foods, fresh fruit, olive oil as principal fat source (monounsaturated fatty acid), low red meat intake, fish (polyunsaturated fatty acid) and red wine in low to moderate amounts. In respect to dietary fats the Mediterranean diet is low in saturated fat but high in monounsaturated fatty acid and polyunsaturated fatty acid.
Diabetes and nutrition
The diabetic diet is essentially a healthy diet. The total fat intake should be less than 35% with saturated fat less than 10%. Total monounsaturated fatty acid 10-20% and polyunsaturated fatty acid less than 10% which corresponds to oily fish 1-2 per week. Interestingly polyunsaturated fatty acid supplements should be avoided in this group as they have been shown to increase LDL-cholesterol. Diabetic patients should also be encouraged to eat regular starchy carbohydrate, avoid sugar, increase fibre, eat regular meals and snacks and avoid diabetic foods.
Cancer and nutrition
Is there a link between diet and cancer?
The World Cancer Research Fund states that 30% of all cancers could be prevented by a change in diet, increased physical activity and healthy weight. Animal studies and metabolic studies have been performed that reveal evidence linking diet and cancer. An ecological study linking diet and cancer compared risk of cancer against intakes of fat, cereals and vegetables in 39 countries (19). The ATBC was a clinical trial that related Vitamin A and E to increased lung cancer rates. (20) 2 large cohort studies have been performed in relation to colorectal cancer and dietary fibre. (21, 22). EPIC showed a significant benefit of dietary fibre against colorectal cancer. The EPIC data showed a 40% difference in colorectal cancer rates between the lowest and highest quintile dietary fibre groups. NIH-AARP however showed no benefit when adjusted for multivariate analysis. The American Gastroenterology Association suggest that available evidence form animal, epidemiological and interventional studies does not unequivocally support protective role of fibre against development of CRC. However the whole body of evidence is analysed overall the overall conclusion is that there is an inverse relationship between dietary fibre and CRC.
Cancer cachexia
Cancer cachexia is defined as anorexia, weight loss and muscle wasting, fatigue and weakness in a cancer patient. Cancer instigates an inflammatory response and production of tumour products. This triggers metabolic abnormalities that produce lipolysis, protein loss and anorexia. As with all disease processes the dietary management of cancer cachexia is initiated by a nutritional assessment. Any symptoms including vomiting and nausea should be treated. There has also been some research regarding fish oils and cancer cachexia. One available product contains docosahexaenoic acid, eicosapentaenoic acid and antioxidants. However a Cochrane review has shown no benefit in relation to beneficial effects of fish oils and cancer cachexia. (23)
Critical care
Injury and sepsis cause major disturbance to clinical state. There is rapid weight loss, increased metabolic rate, protein losses and sodium retention. There are also changes in hormone levels including increased insulin, catecholamines, growth hormone, glucagons and cortisol. The energy requirements in critical care can vary greatly. The Ireton-Jones equation is often used to calculate energy requirements in the critical care setting (See Table 4).
Table 4
Immunonutrition: ESPEN guidelines (34)
Surgical patients
Perioperatively for
Major Cancer Neck Surgery
Major Cancer Abdominal Surgery
Critical Care Patients
Elective Upper GIT surgery
Mild Sepsis
Trauma
ARDS (n-3 FA’s and antioxidants)
Glutamine (burns and trauma)
The specific immune modulating aspects of nutrients have been widely researched in the critical care setting. This field of immunonutrition has shown disparaging results.
Arginine has been investigated as an immune modulating nutrient. Arginine levels can increase or decrease in relation to clinical state. It produces nitric oxide that can lower blood pressure and has been revealed to have detrimental effects in critically ill patients. (24) Glutamine is another possible immune modulating nutrient. It has lots of metabolic functions. It enhances heat shock protein that protect against sepsis. It is thought to be useful in septic shock. Omega-3 fatty acids (alpha-linolenic acid, docosahexaenoic acid, eicosapentaenoic acid) have also been investigated as an immune modulating nutrient. Omega-3 fatty acids produce less proinflammatory eicosanoids than omega-6 fatty acids. There is some evidence that omega-3 fatty acids decrease duration of hospital stay (25) Antioxidants have also been researched as an immunomodulator. Antioxidant levels are lower in critically ill patients. Vitamins A,C,E and Selenium have been studied. A recent meta-analysis has suggested overall mortality benefit but no septic complications benefit in anti-oxidant trials. (26)
Gastrointestinal disease and nutrition
Liver disease
The energy requirements in chronic liver disease are dependent on clinical state i.e. compensated or decompensated. Nutritional requirements for compensated liver disease are 25-35 kcal per kg (dry body weight) day and protein 1.2g per day. Nutritional requirements for decompensated liver disease are energy 35-45 kcal per kg (dry body weight) day and protein 1.5grams per day.
Porto-systemic encephalopathy
This disease process is multifactorial and comprises increased ammonia levels, increased aromatic amino acids, decreased branched chain amino acids and alterations of brain neurotransmitters. There is a widely held belief among doctors that protein intake should be restricted but this is mistaken. Protein requirements are approximately 1g/kg/day and should be divided throughout day.
Ascites
Low salt intake (<6g per day) is an essential component of ascites treatment. Advice should include no salt in cooking, no added salt, avoid processed foods, and avoid foods rich in salt.
Inflammatory Bowel Disease
Inflammatory bowel disease patients are often undernourished due to meagre intake (anorexia, vomiting), amplified losses (diarrhoea and malabsorption) and increased demands (catabolic state). Protein requirements are also increased due to nitrogen losses and catabolic state. It is therefore important that nutritional measures are instituted to improve calorie and protein intake. Interestingly nutrition has been investigated as a treatment for active Crohn’s Disease. Elemental diet is used in active paediatric Crohn’s Disease more than adult Crohn’s Disease to achieve remission. (27). Often this has to be given via naso-gastric tube due to unpalatability. After 4-6 weeks if the patient is in remission foods are introduced slowly over a 3-week period.
Coeliac Disease
Coeliac disease is genetically determined chronic inflammatory disease secondary to gluten (glaidin is the alcohol-soluble fraction) that is a component of wheat. In addition the allergy involves similar proteins found in barley, rye and possibly oats. Coeliac patients consequently exclude these dietary sources. Oats can be reintroduced later depending on response. Dietary sources can be obvious or hidden as gluten can be found in numerous manufactured foods. Coeliac patients can however eat natural gluten-free foods or gluten-free proprietary foods (e.g. Schar, Juvela, Dietary Specials, Glutafin).
Irritable Bowel Syndrome
Simple healthy eating advice is suggested. Diet is tailored to either constipation or diarrhoea symptoms. Typically increasing dietary fibre gradually ameliorates constipation symptoms. Soluble fibre appears to offer benefit more than non-soluble fibre. Decreasing dietary fibre intake treats diarrhoea symptoms. There is not enough evidence regarding exclusion diets although some centres do offer exclusion diets.
Renal disease
Acute renal failure and nutrition is divided into non-catabolic and catabolic patients. Non-catabolic patients do not usually have increased energy requirements. Catabolic patients have high protein requirements but no benefit of >0.2 g nitrogen per kg per day. Their energy requirements should be no greater than >20% above resting energy expenditure.
Chronic kidney disease patients have estimated energy requirements of 35 kcal/kg/IBW/day. (IBW = Ideal Body Weight which in the UK this approximates to BMI 23kg/m2). There has been much research regarding protein restriction and possibility of slowing progression of chronic kidney disease. The Northern Italian Co-op study (protein <0.6grams/day) did show possible slower progression of CKD. However it is known that low protein diets have poor compliance and can increase risk of malnutrition. The Renal Association Standards suggest protein intake 0.75g/kg/IBW/day.
Discussing the dietary management of end-stage renal disease, nephrotic syndrome and renal stones is beyond the scope of this article.
Conclusion
This review article has highlighted the importance of undernutrition in patients under our care. There are numerous methods of screening and assessing patients for undernutrition. There is also a stepwise approach to improving calorie intake: improving oral intake by various methods to enteral and parenteral nutrition. Nutrition is an important aspect of treatment of different disease processes that include cardiovascular disease, diabetes, gastrointestinal disease, renal disease, critical care and cancer. This review article will hopefully provide the medical practitioner with improved knowledge that can be translated into improved awareness and treatment of undernutrition.
Leo Kanner, a Boston physician, first used the word ‘Autism’ in 1943 when he reported on a group of children with deficits in social communication1. Independently, in 1944, Hans Asperger, an Austrian physician, identified similar difficulties in a group of young boys2. Today the term Autism is used to describe a behaviourally defined disorder that is characterised by impairments in social communication, social interaction, and problems with repetitive behaviours and narrow interests.
To receive a diagnosis of autism, a child must have shown delayed language development alongside the characteristic behavioural deficits described in Table One below. Asperger’s Syndrome is used to describe children who had no such delay in acquiring spoken language, and who also have IQ’s above 70. Given that early language development is the key in differentiating between these two disorders, it is not possible for a child to change their diagnosis from Autism to Asperger’s regardless of their later language development and progress.
Debate exists as to whether the two conditions are distinct and it is now generally accepted that they are both part of a spectrum of disorders, hence the term Autistic Spectrum Disorders (ASD). In both the Diagnostic Statistical Manual (DSM IV)3 and International Classification of Diseases (ICD 10)4 Autism and Asperger’s Syndrome come under the category of Pervasive Developmental Disorders. Table One shows the main characteristics of the Pervasive Developmental Disorder.
Table One: Key characteristics of the Pervasive Developmental Disorders
Autism
Deficits in sociability and empathy
Deficits in communicative language
Deficit in cognitive flexibility
Delay with speech development
Detectable before the age of 3
Asperger’s Syndrome
Poor social skills, lack of insight
Behavioural inflexibility, narrow range of interests
IQ over 70
No delay with speech
Motor clumsiness
PDD not otherwise specified
Applies to less severely affected children who do not meet the criteria for either Autism or Asperger’s Syndrome
CLINICAL PRESENTATION
The main characteristics of ASD’s are:
Qualitative impairment in social interaction
Qualitative impairment in communication
Restrictive, repetitive and stereotyped patterns of behaviour, interests and activities
These are known as the ‘triad of impairment’5 and deficits in all three areas must be present for a diagnosis of autism. Each part of the triad will be described, but it is important to remember that not all children with autism will present with all of the difficulties suggested below. Qualitative impairment in social interaction: This includes poor eye contact, poor use of gestures and facial expressions, not sharing, lack of interest in forming social relationships with peers, not joining in with group activities and an inability to recognise the effect of their behaviour on others.
Qualitative impairment in communication: This includes delay in speech, misinterpreting others use of speech such as idioms, sarcasm, jokes and taking things literally. Poor use of speech and also poor understanding of non-verbal gestures such as others’ facial expressions. Limited non-verbal gestures such as pointing.
Restrictive, repetitive and stereotyped patterns of behaviour, interests and activities: Overwhelming interest in a specific topic to such an extent that the child talks about the topic excessively, becomes anxious if unable to perform a ritual or dislikes any interruption to routine and every day life. The child may also have unusual interests such as a fascination with traffic lights, telegraph poles or number plates.
About 70% of children with classic autism have IQ below 708 and approximately one third will have epileptic seizures which continue into adulthood9. PREVALENCE
Recent studies have found a prevalence rate of 20-40 per 10,0006. However if the broader phenotype is used, the prevalence may be as high as 100 per 10,000, or 1% 7. The ratio of males to females is four to one for autism and ten to one for Asperger’s Syndrome. In the last few years, epidemiological studies have suggested that the prevalence of ASD have been increasing. Possible explanations include the fact that the diagnostic criteria has broadened, as well as generally improved case recognition.
ASSOCIATED CONDITIONS
Often in children with autism there are signs and symptoms which are not readily explained by a diagnosis of autism alone. Other medical and psychiatric conditions may co-exist with autism including;
Learning difficulties
Epilepsy
Speech and Language problems
Attention Deficit / Hyperactivity Disorder (ADHD)
Developmental Co-ordination Disorder (DCD)
Tourette’s Syndrome and Tics
Feeding and Eating problems
This is not an exhaustive list8, but we will briefly consider some of the most common conditions and difficulties that a child with autism may also be diagnosed with.
Learning Difficulties: As noted above, approximately 70% of children with classic autism also have an IQ below 70 and are therefore recognised to have mild, moderate or severe learning difficulties9.
Epilepsy: As with learning difficulties, epilepsy is more common among children diagnosed with classic autism, with around 30% being affected into adulthood10. Epilepsy is less common among children with Asperger’s Syndrome, but may be more prevalent than in typically developing children11.
Speech and Language Problems: Most children with an ASD have slower language development than their peers. It is not only expressive language that may show problems, receptive language may also appear delayed in young children, and children may appear to be less responsive to their own name. Some children with autism also appear to lose words that they had previously learnt. This regression is described in approximately 25% of children with classic autism, and is usually a gradual process where a child fails to learn new words, and may stop using previously learnt words altogether12.
ADHD: ADHD is the most common psychiatric disorder to occur alongside an ASD and there are clinical benefits from receiving a dual diagnosis13. Children are likely to benefit from receiving treatment aimed specifically at their ADHD symptoms, as well as having both impairments recognized by parents and teachers.
DCD: Developmental Co-ordination Disorder (or Dyspraxia) describes the motor co-ordination problems and clumsiness typical in AS. Such difficulties may benefit from intervention from an Occupational Therapist or Physiotherapist.
Tics and Tourette’s syndrome: Several reports have documented the co-occurrence of tics in Asperger’s Syndrome. Tourette’s syndrome has also been observed in children with autism. Tics may be verbal or motor. Feeding and Eating Problems: Problems with food including food refusal, selective eating, hoarding, pica and overeating have all been observed among children with an ASD14. Some children have difficulties coping with mixed textures, may eat their food in a certain order and may even ask for their food on different plates.
ASSESSMENT
A general assessment should cover the following areas:
The child’s developmental history.
Observations of the child in structured and semi-structured situations15.
Nursery/School report.
Assessment of cognitive level.
Assessment of problem behaviours.
Speech and language assessment.
Audiology and visual tests if indicated. Chromosomal screen is needed if there are dysmorphic (abnormal) features.
Physical investigations may be specifically indicated in some cases including the need for an EEG, or screening for Fragile X and other chromosomal abnormalities. It is still debatable as to whether these investigations are worth performing routinely as the yield of positive results is relatively low.
Diagnostic Interviews: A number of interviews exist that help clarify the diagnosis and are also used in research. These include the Autism Diagnostic Interview16, the Diagnostic Interview for Social and Communication Disorders17, the Childhood Autism Rating Scale18 and a new computerised interview, the Developmental, Dimensional and Diagnostic Interview (3Di)19. DIFFERENTIAL DIAGNOSES
Information from the above assessments can be used to determine the degree to which a child meets the criteria for an ASD and can also be used to exclude alternative diagnoses. The following conditions should be considered in the differential diagnosis of autism20
- Learning Difficulties
- Hearing Problems
- Speech and Language Disorders
- Rett’s Syndrome
- Childhood Disintegrative Disorder (Heller’s Syndrome)
- Landau-Kleffner Syndrome
- Reactive Attachment Disorder
Learning Difficulties: Children with learning difficulties without an autistic spectrum disorder do not show deficits in their reciprocal social behaviour and their language development is typically in line with their overall intellectual abilities. Hearing Problems: Fluctuating hearing loss, such as glue ear may cause children to show problems in their reciprocal communication, for example, not hearing their name being called. Some may rely on lip-reading during these times of hearing loss, and may appear to make less eye contact. However, these children are capable of making eye contact and may also use sign-based means of social interaction. Speech and Language Disorders: Children with developmental language disorders are unlikely to show the non-verbal communication difficulties typical of children with autism. These children are also less likely to have restricted interests and repetitive behaviours. Rett’s Syndrome: Rett’s Syndrome is a disorder found only in girls. Its typical onset occurs between 5 and 30 months, and is accompanied by a deceleration of head growth. It is characterised by abnormalities in language and social development, as well as a decrease in purposeful hand movements and an increase in stereotyped ‘hand-washing’ movements. Severe or profound intellectual difficulties are also common and epilepsy occurs in the majority of children. Childhood Disintegrative Disorder (Heller’s Syndrome): CDD is characterised by a marked loss of skills following a period of normal development for at least two years. There may also be an increased chance of epilepsy. There is no known consistent cause of CDD. Landau-Kleffner Syndrome: Similarly to CDD, a child with Landau-Kleffner Syndrome would show typical language and cognitive development with a loss of expressive and receptive language skills and seizures consistent with a diagnosis of epilepsy. Landau-Kleffner typically occurs between three and seven years of age and two-thirds of children result in having irreversible receptive and expressive language disorder. Reactive Attachment Disorder: RAD as a result of severe psychosocial deprivation may appear similar to autism in a number of ways. For example, children may have delayed language skills, and may show unusual social interaction and stereotyped behaviours. Early diagnosis may be difficult but once placed in an appropriate social environment, children with RAD tend to gradually develop more typical social behaviours.
AETIOLOGY AND MMR Biological Theories: Genetics play a big role with monozygotic twins of an affected individual having autistic features in 69% of cases compared with zero percent concordance rate for dizygotic twins21. The genetic model is likely to be polygenic in nature with at least 3 to 5 genes responsible. No specific gene has been identified but studies have indicated susceptibility located on chromosomes 2,7, 16 and 17 22.
Imaging techniques have implicated brain regions that play a part in the development of autism including those regions that are responsible for emotional and social functions, regions involving face recognition and social-cognitive systems involved in understanding the intentions of others. A recent fMRI study by DiMartino et al 23 has shown hypoperfusion in the pregenual anterior cingulate cortex in adults with autism. This region is linked to an individual’s capacity to reason about the thoughts and beliefs of others, known as the theory of mind.
The neurotransmitter Serotonin (5-HT) is thought to be involved in autism24. 5-HT is thought to be involved in neurodevelopment and in particular it is abundant in brain limbic areas critical for emotional expression and social behaviour.
MMR: Some parents and families of children with autism believe that the Measles/Mumps/ Rubella (MMR) vaccine caused their children’s autism. These parents’ beliefs and observations were reinforced by a small study of bowel disease and autism, published by Wakefield and his colleagues in 1998 25. The authors suggested that there was a link between the MMR vaccine and autism. However this study was seriously flawed since there was ascertainment bias, unreliable reporting of early symptoms and a lack of a clear pathogenic model.
To date there is no definite, scientific proof that any vaccine or combination of vaccines can cause autism 26. The British Association of Paediatricians recommends that children receive two doses of the MMR vaccine, as long as they have no known health problems that prevent the vaccine from being effective. The immunization schedules recommend that the first dose be given at age 12-to-15 months, while the second dose should be given at either four-to-six years of age or 11-to-12 years of age.
Psychological Theories: Psychological theories have failed to identify one primary deficit that could account for all the features associated with the autistic phenotype.
An interesting theory is the ‘Theory of Mind’ abnormality27. Autistic children lack a ‘theory of mind’and thus are unable to understand that another person can have thoughts, feelings and intentions. MANAGEMENT AND INTERVENTIONS
There is no cure for autism and there is no one specific treatment that is more effective than others (For a review of psychological and educational interventions see Howlin, 199828 and Francis, 200529). However, interventions can be focussed on helping children with autism develop their skills to compensate for their communicative, cognitive and behavioural differences. Interventions need also to be targeted at parents and families to empower them to cope with their children in the most effective way.
Psychoeducation: Receiving a diagnosis of autism is a stressful event for families. The first logical step in providing intervention must be to give parents the opportunity to understand the disorder. Autism is a chronic, life-long neurodevelopmental condition and parents must learn to cope with and manage their child’s behaviours, which may sometimes be distressing and confusing. Children with autism have a lack of empathy, and may not show as much warmth towards their parents as other children. They are also likely to prefer routines, and become frustrated and aggressive if their preferred routine or activity is interfered with. Some children with autism also self-harm. Parental support groups, both national and local, can also offer a much needed source of support and reassurance.
Educational Placement: Improving the child’s educational situation remains one of the most important interventions. While the policy about educational inclusion is somewhat controversial, there is currently no data available about which approach is the most effective, and so choices must be based on pragmatic considerations for the individual. It is sometimes difficult to arrange sufficient support within a mainstream environment, even with a Statement of Special Educational Needs, and so specialist placements may need to be sought. Regardless of the educational placement, structured teaching will help make the school world more comprehensible to a child with autism. The TEACHH programme30, 31 acknowledges the deficits associated with autism and works on structuring learning activities to capitalise on the child’s strengths. For example, children with autism often have good visual processing skills, and so tasks can be structured so that the child can visualise what is expected of them. Special interests can also be used to capture and maintain interest.
Behavioural Treatment: Behavioural analysis of the child’s skills is used to set specific treatment goals and to identify behavioural methods for achieving those goals. Parents as well as other professionals, including teachers and specialist tutors are trained in the implementation of programmes such as ABA (Applied Behavioural Analysis) and Lovaas32. Materials should be matched to the child’s developmental level, and large tasks should be broken down into more manageable tasks to make success more likely. Modelling and reinforcement are key tools in training, helping to increase and maintain desired behaviours.
Some local services and support groups run social skills groups, which can be helpful. If there is a specific behavioural problem then it is helpful talking to a psychologist who can help the parent look more closely at possible precipitants and contributing factors.
Diet: It has been suggested that foods containing gluten and casein may play a role in the difficulties associated with autism33. However, research in this area is scarce so far, and in a recent systematic review34, only one study is considered to be adequate for inclusion35. Based on urine samples, it was suggested that a diet excluding gluten and casein may result in a decrease in autistic traits such as echolalia and rigidity. While this small-scale study and anecdotal evidence may support a diet excluding gluten and casein, such diets are not without their added financial cost and inconvenience, as well as limiting food choices for the affected individual. Further good quality studies are awaited in this area.
Medication: There have been encouraging trials on the use of Risperidone for reducing aggressive and self-injurious behaviour36.
PROGNOSIS
Outcome generally depends on IQ and language development. There may be improvement in language after the pre-school years. However, most individuals continue to show impairments in social skills and communication. Asperger’s Syndrome is associated with a better prognosis due to a relatively greater IQ.
Behaviours and symptoms may vary over time and it is a myth that symptoms remain fixed. Many individuals will require support such help with living independently and obtaining employment. Teenagers may be particularly vulnerable to developing depression and occasionally self-injurious behaviour, particularly if bullying and teasing become a problem.
Chronic infections appear to be common features of various diseases, including neurodegenerative, psychiatric and neurobehavioral diseases, autoimmune diseases, fatiguing illnesses and other conditions.1-4 Neurodegenerative diseases, chronic degenerative diseases of the central nervous system (CNS) that cause dementia, are mainly diseases of the elderly. In contrast, neurobehavioral diseases are found mainly in younger patients and include autism spectrum disorders (ASD), such as autism, attention deficit disorder, Asperger’s syndrome and other disorders.5 For the most part, the causes of these neurological diseases remain largely unknown.2 Neurodegenerative diseases are characterized by molecular and genetic changes in nerve cells that result in nerve cell degeneration and ultimately nerve cell dysfunction and death, resulting in neurological signs and symptoms and dementia.2,3 On the other hand, neurobehavioral diseases are related to fetal brain development but are less well characterized at the cellular level and involve both genetic and environmental factors.6, 7 Even less well characterized at the cellular and genetic level are the psychiatric disorders, such as schizophrenia, paranoia, bipolar disorders, depression and obsessive-compulsive disorders.
Genetic linkages have been found in neurodegenerative and neurobehavioral diseases, but the genetic changes that occur and the changes in gene expression that have been found are complex and usually not directly related to simple genetic alterations.2, 6-8 In addition, it is thought that nutritional deficiencies, environmental toxins, heavy metals, chronic bacterial and viral infections, autoimmune immunological responses, vascular diseases, head trauma and accumulation of fluid in the brain, changes in neurotransmitter concentrations, among others, are involved in the pathogenesis of various neurodegenerative and neurobehavioral diseases.2, 3, 5-16 One of the biochemical changes found in essentially all neurological, neurodegenerative and neurobehavioral diseases is the over-expression of oxidative free radical compounds (oxidative stress) that cause lipid, protein and genetic structural changes.9-11 Such oxidative stress can be caused by a variety of environmental toxic insults, and when combined with genetic factors could result in pathogenic changes.14
Neurodegenerative diseases
Infectious agents are important factors in neurodegenerative and neurobehavioral diseases and may enter the brain within infected migratory macrophages. They may also gain access by transcytosis across the blood-brain-barrier or enter by intraneuronal transfer from peripheral nerves.15 Cell wall-deficient bacteria, such as species of Mycoplasma, Chlamydia (Chlamydophila), Borrelia and Brucella, among others, and various viruses are candidate brain infectious agents that may play important roles in neurodegenerative and neurobehavioral diseases.16-19 Such infections are systemic and can affect the immune system and essentially any organ system, resulting in a variety of systemic signs and symptoms.4, 15, 16, 19, 20
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is an adult-onset, idiopathic, progressive neurodegenerative disease that affects both central and peripheral motor neurons.21 Patients show gradual progressive weakness and paralysis of muscles due to destruction of upper motor neurons in the motor cortex and lower motor neurons in the brain stem and spinal cord. This ultimately results in death, usually by respiratory failure.21, 22 The overall clinical picture of ALS can vary, depending on the location and progression of pathological changes.23
The role of chronic infections has attracted attention with the finding of enterovirus sequences in a majority of ALS spinal cord samples by polymerase chain reaction (PCR).24 However, others have failed to detect enterovirus sequences in spinal cord samples from patients with or without ALS.25-26 In spite of the mixed findings on enterovirus, infectious agents that penetrate the CNS could play a role in the aetiology of ALS. Evidence for transmission of an infectious agent or transfer of an ALS-like disease from man-to-man or man-to-animals has not been found.27
Using PCR methods systemic mycoplasmal infections have been found in a high percentage of ALS patients.28, 29 We found that 100% of Gulf War veterans from three nations diagnosed with ALS had systemic mycoplasmal infections.28 All but one patient had M. fermentans, and one veteran from Australia had a systemic M. genitalium infection. In nonmilitary ALS patients systemic mycoplasmal infections of various species were found in approximately 80% of cases.28 Of the mycoplasma-positive civilian patients who were further tested for various species of Mycoplasma, most were positive for M. fermentans (59%), but other Mycoplasma species, such as M. hominis (31%) and M. pneumoniae infections (9%) were also present. Some of the ALS patients had multiple infections; however, multiple mycoplasmal infections were not found in the military patients with ALS.28 In another study 50% of ALS patients showed evidence of systemic Mycoplasma species by PCR analysis.29
ALS patients who live in certain areas often have infections of Borrelia burgdorferi, the principal aetiological agent in Lyme disease. For example, ALS patients who live in a Lyme-prevalent area were examined for B. burgdorferi infections, and over one-half were found to be seropositive for Borrelia compared to 10% of matched controls.30 In addition, some patients diagnosed with ALS were subsequently diagnosed with neuroborreliosis.31 Spirochetal forms have been observed in the brain tissue of ALS patients and in patients with other neurodegenerative diseases.32 In general, however, the incidence of Lyme infections in ALS patients is probably much lower. In one recent study on 414 ALS patients only about 6% showed serological evidence of Borrelia infections.33 Some Lyme Disease patients may progress to ALS, but this is probably only possible in patients who have the genetic susceptibility genes for ALS as well as other environmental toxic exposures.34, 35
Additional chronic infections have been found in ALS patients, including human herpes virus-6 (HHV-6), Chlamydia pneumoniae andother infections.36, 37 There is also a suggestion that retroviruses might be involved in ALS and other motorneuron diseases.38 McCormick et al.39 looked for reverse transcriptase activity in serum and cerebrospinal fluid of ALS and non-ALS patients and found reverse transcriptase activity in one-half of ALS serum samples tested but in only 7% of controls. Interestingly, only 4% of ALS cerebrospinal fluid samples contained reverse transcriptase activity.39
Although the exact cause of ALS remains to be determined, there are several hypotheses on its pathogenesis: (1) accumulation of glutamate causing excitotoxicity; (2) autoimmune reactions against motor neurons; (3) deficiency of nerve growth factor; (4) dysfunction of superoxide dismutase due to mutations; and (5) chronic infection(s).24, 27-40 None of these hypotheses have been ruled out or are exclusive, and ALS may have a complex pathogenesis involving multiple factors. 28, 36
It is tempting to propose that infections play an important role in the pathogenesis or progression of ALS.28, 40 Infections could be cofactors in ALS pathogenesis, or they could simply be opportunistic, causing morbidity in ALS patients. For example, infections could cause the respiratory and rheumatic symptoms and other problems that are often found in ALS patients. Since the patients with multiple infections were usually those with more rapidly progressive disease,28 infections likely promote disease progression. Indeed, when Corcia et al.41 examined the cause of death in 100 ALS patients, the main causes were broncho-pneumonia and pneumonia. Finally, there are a number of patients who have ALS-like signs and symptoms but fall short of diagnostic criteria. Although a careful study has not been attempted on these patients, there is an indication that they have the same infections as those found in patients with a full diagnosis of ALS (personal communication). Thus ALS-like diseases may represent a less progressive state, in that they may lack additional changes or exposures necessary for full ALS.
Multiple sclerosis
Multiple sclerosis (MS) is the most common demyelinating neurological disease. It can occur in young or older people and is a cyclic (relapsing-remitting) or progressive disease that continues progressing without remitting.42 Inflammation and the presence of autoimmune antibodies against myelin and other nerve cell antigens are thought to cause the myelin sheath to break down, resulting in decrease or loss of electrical impulses along the nerve fibers.42, 43 In the progressive subset of MS neurological damage occurs additionally by the deposition of plaques on the nerve cells to the point where nerve cell death occurs. In addition, breakdown of the blood-brain barrier in MS is associated with local inflammation caused by glial cells.42, 43 The clinical manifestations of demyelinization, plaque damage and blood-brain barrier disruptions cause variable symptoms, but they usually include impaired vision, alterations in motor, sensory and coordination systems and cognitive dysfunction.43
There is strong evidence for a genetic component in MS.44, 45 Although it has been established that there is a genetic susceptibility component to MS, epidemiological and twin studies suggest that MS is an acquired, rather than an inherited, disease.46
MS has been linked to chronic infection(s).46, 47 For example, patients show immunological and cytokine elevations consistent with chronic infections.48-50 An infectious cause for MS has been under examination for some time, and patients have been tested for various viral and bacterial infections. 44, 45,47, 48, 51 One of the most common findings in MS patients is the presence of C. pneumoniae antibodies and DNAin their cerebrospinal fluid.51-53 By examining relapsing-remitting and progressive MS patients for the presence of C. pneumoniae in cerebrospinal fluid by culture, PCR and immunoglobulin reactivity Sriram et al.52 were able to identify C. pneumoniae in 64% of MS cerebrospinal fluid versus 11% of patients with other neurological diseases. They also found high rates (97% positive) of PCR-positive MOMP gene in MS- patients versus 18% in other neurological diseases, and this correlated with 86% of MS patients being serology-positive patients by ELISA and Western blot analysis.52 Examination of MS patients for oligoclonal antibodies against C. pneumoniae revealed that 82% of MS patients were positive, whereas none of the control non-MS neurological patients had antibodies that were absorbed by C. pneumoniae elemental body antigens.53 Similarly, Contini et al.54 found that the DNA and RNA transcript levels in mononuclear cells and cerebrospinal fluid of 64.2% of MS patients but in only 3 controls.
Using immunohistochemistry Sriram et al.55 later examined formalin-fixed brain tissue from MS and non-MS neurological disease controls and found that in a subset of MS patients (35%) chlamydial antigens were localized to ependymal surfaces and periventricular regions. Staining was not found in brain tissue samples from other neurological diseases. Frozen tissues were available in some of these MS cases, and PCR amplification of C. pneumoniae genes was accomplished in 63% of brain tissue samples from MS patients but none in frozen brain tissues from other neurological diseases. In addition, using immuno-gold-labeled staining and electron microscopy they examined cerebrospinal fluid sediment for chlamydial antigens and found that the electron dense bodies resembling bacterial structures correlated with PCR-positive results in 91% of MS cases.55 They also used different nested PCR methods to examine additional C. pneumoniae gene sequences in the cerebrospinal fluid of 72 MS patients and linked these results to MS-associated lesions seen by MRI.56
MRI was used by Grimaldi et al.57 to link the presence of C. pneumoniae infection with abnormal MRI results and found linkage in 21% of MS patients. These turned out to be MS patients with more progressive disease.58 In addition, higher rates of C. pneumoniae transcription were found by Dong-Si et al.58 in the cerebrospinal fluid of 84 MS patients. The data above and other studies strongly support the presence of C. pneumoniae in the brains of MS patients,59-61 at least in the more progressed subset of MS patients.
Other research groups have also found evidence for C. pneumoniae in MS patients but at lower incidence. Fainardi et al.62 used ELISA techniques and found that high-affinity antibodies against C. pneumoniae were present in the cerebrospinal fluid of 17% of MS cases compared to 2% of patients with non-inflammatory neurological disorders. They found that the majority of the progressive forms of MS were positive compared to patients with remitting-relapsing MS. The presence of C. pneumoniae antibodies was also found in other inflammatory neurological disorders; thus it was not found to be specific for MS.62
In contrast to the studies above, other researchers have not found the presence of C. pneumoniaeor other bacteriain the brains of MS patients.63-65 For example, Hammerschlag et al.66 used nested PCR and culture to examine frozen brain samples from MS patients but could not find any evidence for C. pneumoniae. However, in one study C. pneumoniae was found at similar incidence in MS and other neurological diseases, but only MS patients had C. pneumoniae in their cerebrospinal fluid.64 Swanborg et al.67 reviewed the evidence linking C. pneumoniae infection with MS and concluded that it is equivocal, and they also speculated that specific genetic changes may be necessary to fulfill the role of such infections in the aetiology of MS.
Another possible reason for the equivocal evidence linking MS with infections, such as C. pneumoniae, is that multiple co-infections could be involved rather than one specific infection. In addition to C. pneumoniae found in most studies, MS patients could also have Mycoplasma species, B. burgdorferi and other bacterial infections as well as viral infections.68 When multiple infections are considered, it is likely that >90% of MS patients have obligate intracellular bacterial infections caused by Chlamydia (Chlamydophila), Mycoplasma, Borrelia or other intracellular bacterial infections. These infections were found only singly and at very low incidence in age-matched subjects.68 In spite of these findings, others did not find evidence of Mycoplasma species in MS brain tissue, cerebrospinal fluid or peripheral blood.69
Viruses have also been found in MS. For example, HHV-6 has been found at higher frequencies in MS patients, but this virus has also been found at lower incidence in control samples.70 Using PCR Sanders et al.70 examined postmortem brain tissue and controls for the presence of various neurotrophic viruses. They found that 57% of MS cases and 43% of non-MS neurological disease controls were positive for HHV-6, whereas 37% and 28%, respectively, were positive for herpes simplex virus (HSV-1 and -2) and 43% and 32%, respectively, were positive for varicella zoster virus. However, these differences did not achieve statistical significance, and the authors concluded “an etiologic association to the MS disease process [is] uncertain.” They also found that 32% of the MS active plaques and 17% of the inactive plaque areas were positive for HHV-6.70 Using sequence difference analysis and PCR Challoner et al.71 searched for pathogens in MS brain specimens. They found that >70% of the MS specimens were positive for infection-associated sequences. They also used immunocytochemistry and found staining around MS plaques more frequently than around white matter. Nuclear staining of oligodendrocytes was also seen in MS samples but not in controls.71 Using immunofluorescent and PCR methods HHV-6 DNA has also been found in peripheral leukocytes in the systemic circulation of MS patients.72, 73 However, using PCR methods, others did not found herpes viruses in the peripheral blood or CSF of MS patients.74, 75 Evidence that prior infection with EBV could be related to the development of MS was proposed; however, EBV infects more than 90% of humans without evidence of health problems and 99% of MS patients.76 The difference in MS patients could be the presence of multiple infections, including EBV. Recently Willis et al.77 used multiple molecular techniques to examine MS tissue but failed to find EBV in any MS tissues but could find EBV in CNS lymphomas.
Current reviews and the information above points to an infectious process in MS.47, 48, 75, 76, 78-80 Although a few studies did not come to this conclusion,74, 75 most studies have found infections in MS patients. It is interesting that it is the progressive rather than relapsing-remitting forms of MS which have been associated with chronic infections; therefore, infections might be more important in MS progression than in its inception. Various infections may also nonspecifically stimulate the immune system.47, 48 Infections may also invade immune cells and alter immune cell function in a way that promotes inflammation and autoimmune activity.78 If infections like C. pneumoniae and Mycoplasma species are important in MS, then antibiotics effective against these infections should improve clinical status. Although preliminary, that is in fact what has been seen, but not in all patients.81 As in other neurodegenerative diseases, multiple factors appear to be involved in the pathogenesis of MS.
Alzheimer’s disease
Alzheimer’s Disease (AD) is a family of brain disorders usually found in elderly patients and is the most common cause of dementia. AD is characterized by slow, progressive loss of brain function, notable lapses in memory, disorientation, confusion, mood swings, changes in personality, language problems, such as difficulty in finding the right words for everyday objects, loss of behavioral inhibitions and motivation and paranoia. The course of AD varies widely, and the duration of illness can range from a few years to over 20 years. During this period the parts of the brain that control memory and thinking are among the first affected, followed by other brain changes that ultimately result in brain cell death.82
AD is characterized by distinct neuropathological changes in brain tissues and cells. Among the most notable are the appearance of plaques and tangles of neurofibrils within brain nerve cells that affect synapses and nerve-nerve cell communication. These structural alterations involve the deposition of altered amyloid proteins.83, 84 Although the cause of AD is not known, the formation of the amyloid plaques and neurofibrillary tangles may be due to genetic defects and resulting changes in the structure of beta amyloid proteins. This in turn may be caused by chemicals or other toxic events, inflammatory responses, excess oxidative stress and increases in reactive oxygen species, loss of nerve trophic factors and reductions in nerve cell transmission.83-87
Recently AD brain infections have become important.88-90 For example, one pathogen that has attracted considerable attention is C. pneumoniae.91, 92 As mentioned above, this intracellular bacterium has a tropism for neural tissue, and it has been found at high incidence in the brains of AD patients by PCR and immunohistochemistry.92C. pneumoniae has also been found in nerve cells in close proximity to neurofibrillary tangles.92, 93 Similarly to Mycoplasma species, C. pneumoniae can invade endothelial cells and promote the transmigration of monocytes through human brain endothelial cells into the brain parenchyma.94C. pneumoniae has been found in the brains of most AD patients,91 and it has been cultured from AD brain tissue.95 Injection of C. pneumoniae into mice stimulates beta amyloid plaque formation.96 Although the data are compelling, some investigators have not found C. pneumoniae infections in AD.97, 98
AD patients also have other bacterial infections, such as B. burgdorferi.99 Using serology, culture, Western blot and immunofluorenscence methods this Lyme Disease infection has been examined in AD.100, 101 Not all researchers, however, have found evidence of B. burgdorferi in AD patients.102, 103 The presence of intracellular infections like B. burgdorferi in AD patients has been proposed to be a primary event in the formation of AD beta amyloid plaques. This is thought to occur by the formation of “congophilic cores” that attract beta amyloid materials.104 Multiple reports indicate that AD nerve cells are often positive for B. burgdorferi, indicating that this intracellular bacteria could be important in the pathogenesis of AD.99, 100, 104, 105
The hypothesis in AD that intracellular microorganisms could provide “cores” for the attraction of beta amyloid materials is appealing, but other factors, including the induction of reactive oxygen species, lipid peroxidation and the breakdown of the lysosomal membranes releasing lysosomal hydrolases, are also thought to be important in beta amyloid deposition.105 That infections may be important in AD pathogenesis is attractive; however, some negative reports have not confirmed the presence of infections like B. burgdorferi in AD patients.99-101 This suggests that the infection theory, although compelling, remains controversial.102, 105
Herpes virus infections have also been found in AD,especially HSV-1.106, 107 Previously it was determined that HSV-1 but not a related neurotrophic virus (varicella zoster virus) is present more often in AD brains, and this could be linked to AD patients who have the risk factor ApoE e4 allele.108, 109 HSV-1 is thought to be involved in the abnormal aggregation of beta amyloid fragments within the AD brain by reducing the amount of full-length beta amyloid precursor protein and increasing the amounts of their fragments.110 HSV-1 infection of glial and neuronal cells results in a dramatic increase in the intracellular levels of beta amyloid forms, whereas the levels of native beta amyloid precursor protein are decreased.111 This is similar to what has been found in mice infected with HSV-1, indicating that HSV-1 is probably involved directly in the development of senile-associated plaques. Another herpes virus, HHV-6, has also been found in AD patients, but it is thought that this virus is not directly involved in AD pathogenesis. HHV-6 may exacerbate the effects of HSV-1 in AD ApoE e4 carriers.112
Other infections have been found in AD patients, for example, C. pneumoniae, Helicobacter pylori amongst others.113 It has been proposed that such infections may act as a trigger or co-factor in AD.114 Although experimental evidence that pathogens can elicit the neuropathological changes and cognitive deficits that characterize AD is lacking, this approach may yield interesting and important results. These authors also stressed that systemic infections must be considered as potential contributors to the pathogenesis of AD.114
Parkinson’s disease
Parkinson’s disease (PD) is characterized by akinesia, muscular rigidity and resting tremor.103 In addition, autonomic dysfunction, olfactory disturbances, depression, sensory and sleep disturbances and frequently dementia characterize this disease.115 The pathology of PD indicates a progressive loss of the dopamine neurons of the substantia nigra together with the presence of Lewy bodies and alpha-synuclein. More extensive brain degeneration also occurs, from the medulla oblongata to the cerebral cortex.116, 117
Age-related inclusion bodies and protein aggregations or defects in their degradation characteristically occur in PD, but their role in PD pathogenesis remains unclear.117, 118 Some evidence suggests a relationship between PD and specific genetic changes, such as changes in the genes affecting mitochondria, protein degradation, organelle trafficking and vesicular fusion, and in proteins involved in oxidative stress or antioxidant function.102 Inflammation has also been associated with PD pathology.119
The pathogenesis of PD has been proposed to be due to multiple genetic and neurotoxic events that produce oxidative damage and cell death. In the case of PD the relevant targets of toxic events are neuromelanin-containing dopaminergic neurons of the substantia nigra.118, 120 A case-control study indicated that multiple environmental factors and genetic background were statistically related risk factors for PD.121 Prominent among these were long-term toxic exposures and trauma early in life.122 For example,early life exposure to brain injury, chemicals and/or infections may initiate a cyclic inflammatory process involving oxidative damage, excitotoxicity, mitochondrial dysfunction and altered proteolysis that later in life results in substantia nigra neuron death.123, 124
A role for chronic infections in PD pathogenesis has been proposed.123, 124 One infection found in PD that has aroused considerable interest is the presence of chronic gastrointestinal Helicobacter pylori.125 Indeed, treatment of this infection offers relief to late stage cachexia in PD patients receiving L-dopa.126Helicobacter pylori-infected PD patients showed reduced L-dopa absorption and increased clinical disability,127 whereas treatment of this infection increased L-dopa absorption and decreased clinical disability.128H.pylori may not be directly involved in the pathogenesis of PD, but its systemic presence could affect the progression and treatment of PD, probably by stimulating inflammation and autoimmunity.128
Chronic infections in PD have been linked to inflammation and autoimmune responses.129-131 Experimental models of PD have been developed using neurological viral or bacterial infections to initiate the pathogenic process.132, 133 Spirochetes have also been found in Lewy bodies of PD patients.30 Other infections, such as viral encephalitis,134 AIDS-associated opportunistic infections of the basal ganglia,135 coronavirus,136 among other infections,68, 137, 138 have been found in PD and could be important in stimulating inflammation and autoimmune responses. It has been stressed that additional research will be necessary to establish whether a causal link exists between PD and chronic infections.139
Neurobehavioral diseases
Autism spectrum disorders
ASD, such as autism, Asperger’s syndrome, etc., are neurobehavioral diseases of primarily the young where patients generally suffer from an inability to communicate properly, form relationships with others and respond appropriately to their environment. Such patients do not all share the same signs and symptoms but tend to share certain social, communication, motor and sensory problems that affect their behavior in predictable ways. These patients often display repetitive actions and develop troublesome fixations with specific objects, and they are often painfully sensitive to certain sounds, tastes and smells.140, 141
ASD cases are likely to be caused by multiple factors, including genetic defects, heavy metal, chemical and biological exposures, among other important events, which are probably different in each patient. ASD patients appear to have similarities in genetic defects and environmental exposures that are important in patient morbidity or in illness progression.5-8, 140-142
Chronic infections appear to be an important element in the development of ASD.6, 16, 143, 144 In ASD patients more than 50 different bacterial, viral and fungal infections have been found,6 some apparently more important than others in causing symptoms. It has been known for some time that ASD patients have a number of nonspecific chronic signs and symptoms, such as fatigue, headaches, gastrointestinal, vision problems, occasional intermittent low-grade fevers and other signs and symptoms that are generally excluded in the diagnosis of ASD but are consistent with the presence of infections.143 Indeed, increased titres to various viruses as well as bacterial and fungal infections have been commonly seen in ASD patients.6, 16, 19, 143-145 Not withstanding these reports, epidemiological evidence for an association of childhood infections in the first two years of life and ASD has been mixed.146
Environmental exposures to chemicals and heavy metals also appear to be important in the development of ASD.140, 141, 147, 148 The relationship between ASD and heavy metals may involve the role of multiple vaccines in ASD pathogenesis.130, 141 ASD patients often show their first signs and symptoms after multiple childhood immunizations, and the sharp increase in Autism rates occurred only after the multiple MMR vaccine came into widespread use.141 In some states in the U.S. children receive as many as 33 vaccines before they can enroll in school.140 Such vaccines can contain mercury and other toxic preservatives, and some may also contain contaminating bacteria, as found in veterinary vaccines.149
There are very few studies that have followed the transmission of infections and subsequent autism. Previously we found that veterans of the Gulf War with chronic fatiguing illnesses (Gulf War illnesses, GWI) exhibited multiple nonspecific signs and symptoms similar to chronic fatigue syndrome/myalgic encephalomyopathy (CFS/ME).150, 151 After returning to the home with GWI, their children subsequently became symptomatic, and these children were often diagnosed with ASD.152, 153 Symptomatic children (mostly diagnosed with ASD) were infected with the same Mycoplasma species, M. fermentans, that was found in the veterans and their symptomatic family members, and this was not seen in aged-matched control subjects or in military families without GWI. In the GWI families some non-symptomatic family members did have mycoplasmal infections (~10%), but this was not significantly different from the incidence of mycoplasmal infections in healthy control subjects.152, 153
Subsequently ASD patients who were not in military families were examined for systemic mycoplasmal infections.153 The majority (~54%) were positive for mycoplasmal infections. However, in contrast to the children of GWI patients who for the most part had only M. fermentans, the civilian children tested positive for a variety of Mycoplasma species. We also tested a few siblings without apparent signs and symptoms, and for the most part few had these infections.153 In another study we examined the blood of ASD patients from Central and Southern California and found that a large subset (>58%) of patients showed evidence of Mycoplasma infections compared to age-matched control subjects (Odds Ratio=13.8, p<0.001).19 ASD patients were also examined for C. pneumoniae (8.3% positive, Odds Ratio=5.6, p<0.01) and HHV-6 (29.2% positive, Odds Ratio=4.5, p<0.01). The results indicated that a large subset of ASD patients display evidence of bacterial and/or viral infections (Odds Ratio=16.5, p<0.001).19
ASD patients have been examined for B. burgdorferi infections.154 Various studies revealed that 22-30% of ASD patients (N=76) have Borrelia infections.6, 154 The incidence of Borrelia infections in ASD patients may be related to Lyme disease distribution, with some Lyme-intense areas having high prevalence, and other areas having a low prevalence. Other infections, such as Lyme-associated Bartonella, Babesia, Ehrlichia and non-Lyme-associated CMV, Plasmodium species, Toxoplasma species and Treponema species may also be associated with ASD.6
Final comments to part 1
When neurological symptoms are present, infections of the CNS must be considered. Brain infections can stimulate glial responses, and the presence of viral and bacterial infections in nerve cells, can stimulate autoimmune responses against nerve cell antigens as well as the infections within them.155 For example, in MS some 20 different bacterial and viral infections have been found, but the link between these infections and the pathogenesis of MS is still being debated.16,47, 75 One or even a few types of infections cannot be causally linked to MS, and the reason for this is that there may be too many possibilities. No one infection or a group of infections needs to be the trigger in MS to be important in the pathogenesis of MS. In time combinations of certain infections may eventually be identified at least in a subset of MS patients, and this will allow the development of new therapeutic approaches for many MS patients that are not recognized today.
One problem that is rarely discussed is the apparent disparity between the laboratory results from different laboratories. Often different laboratories cannot agree on types of infections found in various chronic diseases.47 There are a number of reasons for this, including differences in the source of materials, qualities of reagents and techniques used.16 Some procedures, such as PCR, have specific challenges that must be overcome in the handling of specimens, their stability, presence of interfering substances, contamination, sensitivity and specificity of the tests and interpretation of the results. Variability in results from different laboratories will remain a problem unless research groups work closely together to solve these problems. One example of how this has been overcome is a multi-centre research study on the presence of C. pneumoniae in the cerebrospinal fluid of clinically defined, mono-symptomatic MS patients.156Sriram et al.156 conducted this diagnostic trial with good concordance of results between different laboratories. Cooperative studies such as this should eventually alleviate discrepancies in the types of infections found by different research groups.
This review continues in Part 2 with psychiatric diseases, autoimmune diseases, fatiguing illnesses, and other infectious diseases with neurological aspects and an overall discussion of the topic. 157
Introduction:The aim of the NHS Modernisation Agency’s Hospital at night project is to “Redefine how medical cover is provided in hospitals during the out of hours period.”(1) The project “requires a move from cover requirements defined by professional demarcation and grade, to cover defined by competency in order to release significant amounts of medical staff time and support the compliance with WTD (Working Time Directive) while enhancing clinical practice and training.”(1) Chairman of the BMA’s Junior Doctors Committee and also medical advisor to the Hospital at Night Project undertook a survey of junior doctors’ activity in the evening, night, and weekends. They noted that the evenings were very busy and the nights quieter, that general physicians work harder at night than those in other specialties or orthopaedic surgeons. In summary their survey showed that a huge amount of doctors’ time was wasted on tasks which did not require their level of skills. Such tasks included phlebotomy, searching for notes, and x-rays.(1) An evaluation report into the implementation and impact of the hospital at night pilot project, August 2005, identified key elements to minimize medical workload at night. These included working within a multidisciplinary, competency-based team, with extended skills ward staff to minimize reliance on the night team, reduce duplication, take away inappropriate tasks, bleep-filtering and better use of new technologies such as digital imaging and e-prescribing (2). Our aim was to assess the experience of a typical junior orthopaedic doctor’s experience during his on-call to assess their perspective of the implementation of hospital at nightand to establish whether the recommendations of the evaluation report were being implemented. Methods: A prospective review was conducted of the night duties of all junior doctors working or cross-covering trauma and orthopaedics across Frenchay and Southmead hospital sites of the North Bristol NHS Trust between December 2007 and January 2008.A questionnaire included nature of activity and level of experience, closed questions relating to the hand-over experience. Details of calls received and the nature of tasks undertaken during on-call period with times. Data were then stored and analysed using excel-office 2002. Results:Total of 51 questionnaires were completed by the junior orthopaedic doctors during their respective on-call duties. A total of 109 calls were received or tasks requested. The average time spent during hand-over was 14.1 minutes (range 5-20 minutes). Discussion: Our data indicates busiest times occurred between 21:00 – 23:00 (Fig 1) Figure 1, illustrates a distribution of the peak work-load period which occurred between 9pm-11pm and 1am-3am. This finding contradicts previous surveys done prior to the implementation of the hospital at night where it was noted that the nights were quieter and sub-specialist such as orthopaedic surgeons were not working as hard as general medics (1). Figure 2, shows the overall experience of the hand-over as judged by the orthopaedic juniors. There was an eighty-five percent (43/51) attendance by the on-call orthopaedic juniors of which seventy-two percent (32/44) thought that it was a useful experience Figure 3 illustrates a distribution of the calls received. As illustrated, 37/109 calls were viewed as inappropriate The majority of the orthopaedic juniors surveyed framed hospital-at-night hand-over was useful but thirty-four percent (37/109) of the calls they received were viewed as inappropriate (Fig 2,3). The most frequent of the inappropriate ward calls were ‘requests to rewrite drug-charts’ (Fig 4,5). Figure 4 describes the frequencies of these calls. As illustrated the most frequently inappropriate ward-call was to ‘rewrite drug-charts’. There were twenty-three (23/37) inappropriate ward-calls Figure 5 shows the reasons given by the on-call doctors for the inappropriate tasks. The most frequently given reason was that ‘the request should have been done during the day’. The second most frequently given reason for inappropriate calls was the fact that ‘it was a duplicated request’ Our survey also highlighted that the most frequently given reason for inappropriate tasks was firstly that the request should have been done during the day and secondly that it was a duplicated request. These findings seem to defeat the key purpose of the hospital-at-night project for an orthopaedic SHO on-call. Conclusion: Hospital at night cannot function in isolation. There are fewer doctors available then normally and the system has to use their time effectively. 30% of inappropriate calls to wards were to rewrite drug charts and to prescribe warfarin. This should have been identified and performed by the day staff. 10% of calls were to review X-rays. Much of the inappropriate activity could be pre-empted if both day and night staff attended the handover and the tasks outstanding from the day identified. The doctor on the night could then perform an hourly ward round completing these tasks without interrupting at the beginning of the night.
Introduction Dysmenorrhoea and other premenstrual symptoms are common among women of reproductive age and lead to suffering and impact on home, school, and work performance. Earlier studies have focused on the prevalence and risk factors of dysmenorrhoea 1, 2, 3. Surveys in Pakistan have found the prevalence of premenstrual symptoms varying from 53% to 67% in college girls 4, 5, 6. About 57% of students in one study reported that dysmenorrhoea affected their work5. Although these surveys document the prevalence and severity of symptoms they do not correlate it with the impact of specific symptoms on daily activities or with healthcare seeking behaviour. A population-based survey of 2262 women from Goa, India, revealed a linear association between pain severity and treatment seeking and time off from work 3. However the impact of specific symptoms of premenstrual syndrome on treatment seeking and rest was not reported. Aims Primary objective: The authors carried out a cross-sectional study to explore the impact of dysmenorrhoea and other premenstrual symptoms among women of reproductive age.Secondary objective: The authors intended to find out the predictors of healthcare-seeking behaviour including self-treatment for premenstrual symptoms. Methodology A cross-sectional survey study design was chosen. A 13-item questionnaire was administered to women, aged 16 to 50, who were not pregnant and had no known gynaecological, medical, musculoskeletal or neurological diagnosis. Participants were recruited from multiple sites using convenience sampling within urban areas of Islamabad and Rawalpindi in Pakistan. Trained interviewers (physicians, medical students, nursing staff, and high school students) filled out questionnaires interviewing female students at a medical college, a nursing college, and at city schools; as well as housekeeping staff and patients’ attendants at a tertiary care teaching hospital in Islamabad. A proportion of women completed the questionnaire themselves. Ethical approval was obtained from the Shifa International Hospital Ethics committee. No personally identifiable data such as the respondents’ names were recorded. ResultsStudy population characteristics A total of 1236 women from multiple locations within Islamabad and Rawalpindi, Pakistan, participated in the survey. Most of the women were in the younger age groups: 402 women (33%) were 16–20 years old and 622 (50%) were 21–35 years old. Fewer women were in the older age groups: 147 women (12%) were 36–45 years old and 63 (5%) were more than 45 years old. About 55% were unmarried and 61% had no children. The distribution of educational achievement was weighted towards the more educated: 16% were able to read a religious book (basic literacy); 49% had some school education (up to 12th grade); and 34% had professional level education. About 27% of the respondents were homemakers (among women with professional level education, 10% reported staying at home). Age at menarche was less than 12 years old for 16%; between 12 and 14 years for 62%; and more than 14 years old for 23% of the respondents. Severity of dysmenorrhoea and its correlation with premenstrual symptoms On the 10-point visual pain scale, 465 women (38%, 95% CI 35–41) reported mild dysmenorrhoea severity from 0 to 3; 517 women (42%, 95% CI 39–45) reported moderate severity from 4 to 7; and 248 women (20%, 95% CI 18–23) reported severe pain rated from 8 to 10. The linear regression coefficient between dysmenorrhoea severity score and the number of days unable to work in a month was 0.59 (standard error: 0.031). Table 1: Prevalence of premenstrual symptoms and their contribution to pain severity and days lost from work
Women reporting symptoms, n (%, 95% CI) a
Contribution to pain score b
Contribution to days out of work c
Low back pain
879 (72, 69–74)
0.39 *
0.05
Depressed mood
484 (40, 37–43)
0.17 *
-0.05
Headache
268 (22, 20–24)
0.25 *
0.22 *
Swelling
218 (18, 16–20)
0.03
0.12
Nausea
218 (18, 16–20)
0.17 *
0.01
* Statistically significant values (P < 0.005)a Respondents were allowed to select more than one option. Total respondents: 1236.b Linear regression coefficients for a 10-point visual pain score categorized into three levels.c Linear regression coefficients for self-reported days unable to work in a month. Table 1 shows the prevalence of primary symptoms of preceding menstruation and their contribution to a 10-point visual pain scale and to self-reported days unable to work in a month. Low back pain and headache contributed most to the pain score while headache and swelling correlated with days out of work. Impact of dysmenorrhoea Among the working women (366 professionals and housekeeping staff) 49% (95% CI 44–54) reported one or more days out of work in a month due to pain. Similarly, among the 452 students surveyed, 53% (95% CI 48–58) reported dysmenorrhoea affecting school performance (Table 2).
Table 2: Impact of premenstrual symptoms and dysmenorrhoea on household, school and work performance
All respondents, n (%)
Students, n (%)
Maids and housekeeping staff, n (%)
Self-employed, n (%)
Professionals, n (%)
Affected domain
Household chores
441 (37)
124 (28)
59 (40)
31 (52)
75 (35)
Household income
129 (11)
24 (5.3)
33 (22)
12 (20)
18 (8.5)
Performance in school
313 (25)
239 (53)
N/A
N/A
N/A
Social obligations
395 (33)
130 (29)
58 (39)
31 (52)
85 (40)
Unable to work for one or more days in a month
643 (53)
282 (63)
75 (51)
38 (63)
103 (50)
Percentages do not add up to 100% as respondents were allowed to select more than one affected domain.Values for 50% or more respondents acknowledging an impact in a domain are given in bold.
Treatment taken for dysmenorrhoea relief
Table 3 outlines the remedies sought for the relief of dysmenorrhoea symptoms and the reported effectiveness of each type of remedy. All treatments except homeopathic were felt to be effective. Logistic regression analysis showed that the use of any treatment type was related to low back pain (odds ratio 2.2, 95% confidence interval 1.6–2.9), pain severity (OR 2.0, 95% CI 1.6–2.5), headache (OR 1.7, 95% CI 1.2–2.4), depressed mood (OR 1.7, 95% CI 1.3–2.2), increasing education (OR 1.1, 95% CI 1.0–1.2) and not being unmarried (OR 0.52, 95% CI 0.34–0.79).
Table 3: Remedies sought for dysmenorrhoea relief and their patient-reported effectiveness
Women reporting use of treatment, n (%, 95% CI)a
Odds ratio for self-reported effectiveness, (95% CI)b
Conventional medicine
496 (56, 53–59)
13 (8.7 to 21)
Household remedies
285 (32, 29–35)
6.5 (4.1 to 11)
Herbal
90 (10, 8–12)
4.1 (2.2 to 7.7)
Homeopathic
125 (14, 12–17)
1.5 (0.89 to 2.6)
a Respondents were allowed to select more than one option. Total respondents: 1236. b Unconditional logistic regression (converged, 6 iterations, 878 cases included, using Epi Info 3.4.3). All odds ratios with P < 0.005, except homeopathic, P = 0.12.[CI, confidence interval]
Conventional medicine was sought by women with higher pain scores (OR 2.2, 95% CI 1.8–2.8) and greater years of education (OR 1.2, 95% CI 1.1–1.3); other factors such as age and specific symptoms were not statistically related. Women with a greater number of days unable to work were more likely to use herbal treatment (OR 1.4, 95% CI 1.1–1.9) while household remedies like hot water bottles and warm drinks were more commonly taken by women with headache (OR 1.66, 95% CI 1.2–2.4), depressed mood (OR 1.6, 95% CI 1.2–2.3) and lesser years of education (OR 0.88, 95% CI 0.78–0.99). Discussion This paper is the first, to the best of our knowledge, to show an association between specific premenstrual symptoms and dysmenorrhoea severity and healthcare-seeking behaviour. We found that certain symptoms, namely low back pain and headache, contributed more to the perceived severity than other complaints. This finding may be of importance to clinicians treating women with menstrual complaints especially when the treatment is symptom-oriented. In women working outside their homes clinicians may wish to target headache and swelling as these symptoms correlated with days unable to work. Prevalence of premenstrual symptoms in the present study was higher than a Japanese survey7 that reported back pain in 6.9% of women and headache in 11% of women (compared with our results of 72% and 22% respectively). This difference may be due to cultural differences in perception and reporting of symptoms, overall better health, and being strong and hardworking. It may simply be the perception difference in underdeveloped or developed country i.e. lack of resources, poor diet, and poor health. Menstrual symptoms caused a heavy impact on social, school, and work responsibilities in women, a finding we share with previous studies. A cross-sectional survey from India found that 17% of adolescent girls reported missing school classes due to dysmenorrhoea while 60% reported disruption of their daily activities8. In an Australian study, 53% of high school girls reported that dysmenorrhoea limited daily routines and 37% stated that it affected schoolwork9. A study from New York found 46% of students missing one or more days of school due to dysmenorrhoea10. Corresponding figures from the present study were: 62% students reported missing at least one day of school and 53% reported an impact on school performance. The authors would like to reiterate the need for screening for and treating menstrual symptoms because of the impact on daily activities and the potential to reduce avoidable suffering. Women seek a variety of sources for relief of menstrual symptoms. A survey of 2411 high school girls in Malaysia showed that 11% sought medical care although the majority (80%) obtained advice from their mothers regarding premenstrual symptoms11. A study of adolescent girls in Haryana, India, found that 5.3% consulted a physician for menstrual symptoms and 22% self-treated with over-the-counter medicines12. 52% reported self-treatment and 7.7% used complementary medicines in a Japanese study13. In the present study, although conventional medicine was felt to be most effective, it was used by only half of the women. This indicates poor access or awareness of available effective treatments. Even household and herbal remedies were infrequently used possibly due to their limited effectiveness. As menstruation and its associated symptoms are often thought to be a ‘normal’ part of women’s lives these issues may remain untreated in the community. Women who were more educated tended to seek more effective treatments, as did those who had more severe symptoms. Clinicians and public health professionals need to proactively reach women from less privileged background to reduce suffering from menstrual symptoms. Educational campaigns to improve awareness of safe and effective conventional medicine, such as non-steroidal anti-inflammatory drugs, could reach women not aware of these options. These public health campaigns may be addressed toward high-school girls, homemakers, and professionally employed women through separate targeted channels. The present study was limited by non-random (convenience) sampling yielding a study sample skewed towards more educated women. This may be due to sampling in urban areas only. The questionnaire was designed using closed-ended questions, to reduce subjectivity in data recording, limiting exploration of unanticipated variables. Psychosocial issues, such as socio-economic disadvantage and mental health, play an important role in the perception and reporting of menstrual symptoms3 but these factors were not explored in this study. Further research in this area should focus on awareness, access to care, and quality of life outcomes with different treatment options. ConclusionLow back pain and headache contribute the most to severity of dysmenorrhoea while headache and body swelling (fluid retention) were predictive of days unable to work. Conventional medicine is commonly used by more educated women, as well as those with more severe symptoms, and was perceived to be effective more often than other treatment modalities. Effective treatments for the relief of menstrual symptoms remain underutilized causing avoidable suffering.
IntroductionHospital inpatient falls have remained the subject of extensive research and intervention over the past 55 years1. Despite risk factor identification, multiple prediction system developments and harm reducing technologies, the issue of falls remains. The incidence of falls varies between 2.2 and 14 per 1000 patient days 2,3 and increases with age 4. In the UK’s National Health Service, 32% of adverse incident reports are due to falls 2 . In 2007 it reported 200,000 inpatient falls to the National Patient Safety Association 5. Whilst most patients (96%) came to no harm or minor harm, they estimated that over 500 hip fractures and 26 deaths resulted from these falls.Inpatient falls lead to greater morbidity and mortality than equivalent fractures in the community, 6 and they significantly increase the length of stay in hospital7. Apart from physical harm, there are also psychological consequences for patients such as anxiety, loss of confidence, and fear of falling 4. The risk of falls leads to a conundrum in rehabilitative care. Ideally, patients’ mobility, autonomy and dignity must be encouraged and respected - yet slips, trips and falls must be pre-empted often with varying degrees of intrusion and even restraint. In order to selectively target individual patients who would benefit from closer attention, many risk stratification tools have been developed to predict potential fallers. They were developed on a background of over 400 independent risk factors identified with falls 8. The most prominent in the UK are STRATIFY (St Thomas's risk assessment tool in falling elderlyinpatients) and Downton with the recent addition of the Wandering Behaviour Assessment 8. STRATIFY 4 was designed to be used once per week and assesses 5 factors –falls history, patient agitation, visual impairment that limits daily function, frequent toileting, and a transfer or mobility score of 3 or 4 . Each factor scores 1 point and a score greater than 2 was found to have a 92% sensitivity and 68% specificity for a fall in the following week. In the Wandering Behaviour study the STRATIFY criteria results were not reproduced and were found to have only an 82% sensitivity and 34% specificity in their study population 8. Downton 9 also has 5 categories of assessment; falls history, medication subtypes, audio-visual-sensory deficits, mental state (using the Mini Mental State Score <24) and stability of gait. Each category scores 1 point and a score of greater than 3 is considered significant. In the Wandering Behaviour study, when applying the Downton criteria they only found an 82% sensitivity and 36% specificity 8. The Wandering Behaviour assessment looks for the presence of the following :checking, pottering, aimless walking, walking with inappropriate purpose, walking with appropriate purpose but inappropriate frequency, excessive activity, night-time walking, attempts to leave the hospital and being brought back to hospital. The presence of any one of these was found to have a sensitivity of 43% but specificity of 91% 8. Thus, both STRATIFY and Downton are limited by the multi-disciplinary assessments required, so that even when implemented, it is difficult to repeat them on a daily or weekly basis. The Wandering Score is easily repeatable on a daily basis, but lacks the sensitivity of STRATIFY and Downton 8. The aim of this study is to investigate whether changes in a patient’s physiology can be predictive of falls risk, and if so, can they become a useful tool for calculating risk? The reason why we have chosen to investigate this is that many of the commonly encountered risk factors can potentially be reflected in the patient’s routine observations 10. For example, fever could indicate infection and delirium. Hypotension could result from anti-hypertensives, sedatives, or dehydration. Hypertensive states could result from stroke, stress response to infection, or even be a surrogate marker of cardiovascular disease predisposing to arrhythmias. Blood pressure variation could also be predictive since dynamic orthostatic challenges such as lying and standing blood pressure measurements are known to predict falls 10. Thus, by measuring fluctuations in the observations we hypothesised that it may be possible to make an accurate short term prediction of falls riskMethodsSubjectsWe obtained consent for this retrospective study from the hospital’s ethics committee. We based this study in the Acute Medical Unit of our hospital, as it has the largest case mix of patients recently admitted and is therefore most likely to have the largest physiological instability. We aimed to detect a mean difference of 10% in the physiological variability prior to falling with 80% power, p-value < 0.05, and a common standard deviation of 5 %. Our power calculation indicated that we needed to study 12 patients. We identified all the falls in the calendar year of 2008 by examining the records of all the incident report forms submitted by the ward. A total of 33 incident reports related to falls were logged. Two reports related to staff slips and trips and were excluded, and a third incident report failed to adequately identify either the patient’s name, date of birth or hospital number and so had to be excluded. One of the 30 patients fell twice, and this was treated as its own incident as with the STRATIFY paper. The incident report forms were also used to identify the time, nature, and outcome of the fall. Of all the case notes related to the 30 falls requested from medical records, only 13 were available for detailed review. MeasurementsHeart rate (HR), Blood Pressure (BP), Temperature and Respiratory Rates were all recorded and analysed. The PAR (Patient at Risk) Score as calculated by the nursing staff was included too. PAR Scores are validated mechanisms of identifying sick patients who may go on to develop deterioration to the point of requiring Intensive Care. Scores of >3 are associated with a high risk of deteriorating health and are calculated using the routine nursing observations set. Oxygen saturations were not included in this study, as even small deviations tend to be rapidly corrected by staff with oxygen and therefore were not thought to be a useful marker. Recordings of blood pressure and heart rate were all done with ward based equipment. It is impossible to know which machines were used on individual patients as the equipment has varied, both over the course of time, and between multiple wards. However, because the subjects were their own controls, we are confident that the same machine and cuff were used for taking all the 12 hour recordings as each individual machine is allocated to a given bay. Blood pressures were obtained using semi-automatic Dynamap equipment, therefore the readings are in effect calculated from the Mean Arterial Pressure (MAP). We elected to study the systolic readings only, as this represents the maximum perfusion pressure to the brain and carotid sinus. Two of the thirteen patients were known to have undergone orthostatic challenges as part of their admission work up and these were included. Other parameters recorded included the time of the fall, number of medications on the drug chart, whether the fall was observed and any injuries sustained. ControlsWe had two sets of controls. The first set was used to compare the age and gender profile of the fallers to those of the general adult admissions. We had to do this because the official hospital statistics included obstetric and paediatric admissions and comparison would have been inaccurate. We therefore took 4 random days’ of acute adult medical admissions (i.e. over the age of 18) in order to compile an age and gender profile of patients admitted with a ratio of 4:1 (n=110). In order to compare the variability in physiological parameters between fallers and non fallers we generated a second set of controls matched for age (± 5 years) and gender in a ratio of 4 to 1 from a random week’s cohort of patients in February 2009 (for practical purposes). For each matched control, recordings were made at the equivalent length of stay and the controls were not known to have previously fallen. The patients were taken from across the hospital’s medical wards (not Intensive Care or Surgical) with their diagnosis blinded from the investigators. We were able to match 47 of the 52 controls that we were aiming for. Only BP and HR data were recorded in the controls, as we already established that temperature and respiratory rate were not sensitive markers from our faller data. StatisticsOur 13 patients allowed us to reach sufficient statistical power of 80%. Physiological parameters were tested within groups using the Students t-test (paired, two tailed) and when comparing to controls using the Students t-test (unpaired, two tailed). The null hypothesis were rejected when p<0.05. Variability in heart rate, temperature, respiratory rate and PAR (Patient At Risk Score) were calculated using the mean, range and standard deviation between maximum and minimum values. Blood pressure variability was measured by using the maximum and minimum systolic pressure and calculating the mean, range and standard deviation. We have chosen to primarily measure variability by the range as it is a simple calculation that can be done by anyone on a ward. It doesn’t require a calculator nor any detailed knowledge of mathematics. It is therefore an effective and repeatable measure which could be easily implemented as part of a scoring system. Data processing and Statistical Analyses was done in MS Excel 2002 and SPSS v.14. ResultsThe mean age of fallers was 79 years old (SD 16, n=30) and of the acute medical admission controls 67 years old (SD 20, n=110) with p=0.003. The gender distribution of males to females was 63% to 37% respectively for fallers (n=30), but 38% to 62% in general medical take controls (Fishers exact test p<0.001). 54% of fallers (n=13) were admitted from their own home with 31% from Residential Homes and 15% from Nursing Homes. 54% were known to have a preceding falls history (n=13). The timings of the falls showed that 60% of falls occurred between 08.00 and 20.00 (n=30). 77% of falls occurred within 48 hours of admission (n=13). The circumstances of the falls were consistent with the NPSA statistics with 20% falling out of bed, 17% from chair, 17% from commode / toilet, 20% when walking, 3% in the bath and 23% not documented. The significance of the falls as measured by the incident report forms (n=26, in 4 cases not recorded) was 70% under the level of 6 (i.e. low level and did not require further investigation), 13% were over the level of 6 (i.e. were serious and required further investigation) and 17% were unrecorded. Most of the injuries (n=30) were none or minor – 47%, 10% had a head injury and 43% were not recorded. 95% of the falls were not observed. 31% of the patients (n=13) were taking fewer than 4 medications, 38% were taking between 4 and 7 medications and 32% were taking over 8 medications. Comparing the demographic characteristics of our fallers to the age and gender matched controls; average age was 79 for both fallers and controls (p=0.94) and the gender distribution was 7 females : 6 males in the fallers, and 27 females: 21 males in the controls (Fisher’s Exact Test p=1.0). The overall PAR score (n=13) was not sufficiently sensitive to predict falls risk – 77% had no change in their PAR score, 8% had a 1 point change and 15% had a 2 point change ( Table 1). Temperature variation (n=13) was minimal with 69% having less than 1 degree Celsius change and 31% having change of 1 to 1.5 degrees Celsius . Recorded respiratory rate variation was also minor (n=13) with 85% having a maximum change of up to 4 breaths per minute. Table 1. Physiological Parameter Data * denotes p<0.05
Fallers N = 13Mean ± SDRange (x-x)
Controls N = 47Mean ± SDRange (x-x)
PAR Score
1.2 ± 0.6(1 – 3)
PAR Score Variability
0.4 ± 0.6(0 – 2)
Temperature (Celsius)
37.1 ± 0.5(36.4 - 38.1)
Temperature Variability (Celsius)
0.6 ± 0.5(0 – 1.3)
Respiratory Rate (Breaths/Minute)
18.5 ± 1.9(16 – 22)
Respiratory Rate Variability (Breaths/Minute)
1.1 ± 1.5(0 – 4)
Highest Heart Rate (Beats/Minute)
86 ± 13(70 - 110)
86 ± 14(60 -112)
Lowest Heart Rate(Beats/Minute)
71 ± 16(50 -101)
79 ± 14(55 – 110)
*Heart Rate Variability (Beats/Minute)
15 ± 10(0 – 39)
7 ± 7(0 – 27)
Highest Systolic BP (mmHg)
142 ± 22(102 – 180)
140 ± 24(99 – 218)
Lowest Systolic BP(mmHg)
116 ± 19(71 – 150)
129 ± 24(86 – 199)
*Systolic Variability (mmHg)
26 ± 12(2 – 40)
11 ± 7(0 – 26)
In fallers (n=13), the mean highest heart rate was 86 bpm (SD=13) and the mean lowest heart rate was 71 bpm (SD=16). The range was 15 bpm (SD=9.6) with p<0.001. In controls (n=47) the mean highest heart rate was 86 bpm (SD=14) and the mean lowest heart rate was 79 bpm (SD=14). The range was 7 bpm (SD=7) with p<0.001. The significance test when comparing the highest average heart rate between fallers and controls shows p=0.98 showing that they were well matched. The significance test for comparing the variation of the heart rate between fallers and controls is p<0.001. In fallers (n=13), the mean highest systolic BP was 142 mmHg (SD=22) and the mean lowest systolic BP was 116 mmHg (SD=19). The average variation was 26 mmHg (SD=12) with p<0.001. In controls (n=47) the mean highest systolic BP was 140 mmHg (SD=24) and the mean lowest systolic BP was129 mmHg (SD=24). The average variation was 11 mmHg (SD=6.5) with p<0.001. The significance test when comparing the highest average systolic BP between fallers and controls shows p=0.77 showing that they were well matched. The significance test for comparing the variation of the systolic BP between fallers and controls is p<0.001. Charts 1 and 2 show the spread of measurements in the cardiovascular parameters between fallers and controls.
Chart 1. Comparison of the heart rate variation between Fallers and Controls
Chart 2. Comparison of the systolic blood pressure variation between Fallers and Controls
DiscussionThe average age of the fallers was significantly greater than the average age of those admitted to the hospital in general. This is unsurprising considering both mechanical deterioration of the musculoskeletal system with advancing age and the accumulation of disease processes. It is noteworthy that despite making up the smaller percentage of admissions to the hospital, men made up the greater proportion of fallers. Other much larger studies show considerable variability in their gender proportions 11,12, therefore this finding is unlikely to be truly significant. Apart from age and gender, polypharmacy was also a feature of our fallers. This is consistent with other studies 13,14,15. Additionally, our data was consistent with the overall NPSA statistics in terms of the significance and circumstances of the falls 5. The main and novel finding of our study was that fallers were significantly more likely to display a larger range in their cardiovascular observations than the standard hospital population. Whilst it is generally expected that subjects undergoing any routine measurement of heart rate and blood pressure will have a variation in measurements of about 10% over the course of 12 hours 16,17, we found that our fallers had a variation in their heart rate and blood pressure of approximately 20%. This is similar to the dips experienced by the normal population over the course of the night and during orthostatic challenge. This was despite the fact that the baseline measurements of highest value were virtually the same for both populations. Furthermore, almost all the values recorded were within normal limits – and would not normally require specific remedial action to be taken. This could also explain why this risk factor has not previously been identified in other studies. Our study indicates that it is the cardiovascular lability rather than the cardiovascular measurements per se, which acts as an acute predictor of falls. In fact, the sensitivity for falls prediction with either a range of HR values > 15 beats per minute or range of BP systolic values >25 mmHg was 77%. We would therefore expect that when such patients mobilize, the superadded orthostatic challenge would be too great for cardiac output to be suitably matched and so patients are at greater risk of falling. In terms of the 12 hour prospective risk of falling this could certainly explain why a patient with known risk factors will fall during a given nursing shift. Indeed it may also explain why a patient may fall during a hospital admission when patients were, for example, already parkinsonian and arthritic and yet had not previously fallen. Interestingly, neither temperature, respiratory rate, nor PAR Score showed any significant lability in the lead up to the falls. This was surprising as we would have expected them to be predictive of other well known risk factors. The lack of fluctuation in temperature and respiratory rate could provide further evidence that the key short term factor responsible for falls is cardiovascular lability. More detailed analysis showed that it was more likely that these measurements were insufficiently sensitive. Only four of the fallers were admitted with infections, and those subjects showed some temperature fluctuations. However, only 2 had temperatures above 37.5 degrees Celsius, which is consistent with the blunted fever response that is well known to occur in 50% of the elderly population 19,20 (and most fallers were elderly). The lack of value in respiratory rate recordings probably reflects the lack of due care and attention paid to this, the only manually measured parameter. It has long been recognized that respiratory rate recordings tend to be inaccurate 21 . The highly limited range of measurements recorded (16-22 breaths per minute) amongst all the fallers, despite some patients having severe pneumonia, further supports this finding. Finally, the PAR score tended to be quite static. This was a result of its constituent parameters not being sensitive (temperature, respiratory rate) and the fact that most of the heart rate and blood pressure recordings were within normal limits. LimitationsDespite the fact that this study was well powered and statistically significant, ultimately it is quite limited in numbers with just 13 patients. It was disappointing that we were not able to obtain case-notes or the appropriate file in the other 17. We are also presenting calculated data from the MAP measurements, without knowing the exact algorithms being used. For this reason, we analysed the given systolic pressures as further data manipulation would have increased inaccuracies. Our data is taken from relatively acute admissions and as such may not necessarily be applicable to long stay patients, where cardiovascular lability may not play an important role.Furthermore, controls were not matched for diagnosis or for the number of medications taken. This could lead to criticism that the comparison was poor, though the baseline measurements for the two were remarkably consistent. One of our aims was to see if variability could be used to accurately model general falls risk. We therefore thought it would be more useful to study the hospital’s general physiology, in all its varying degrees of illness. ConclusionsThis study shows the value of looking closely at patients’ observations and that even ‘normal’ values have to be interpreted in context . The data supports the finding that the risk of falling at a given point in time relates not only to predisposing factors, but also to their current cardiovascular status. We therefore suggest that a one-off falls risk assessment is no longer appropriate, but should be continuously reviewed on a shift-by-shift basis by nursing staff. This has significant ramifications for modernizing current risk stratification tools so that they are able take this into account.
Barrett’s Oesophagus (BO) describes a histological abnormality of the lower oesophagus widely accepted to be associated with gastro-oesophageal reflux disease (GORD). The nature of this disease has been a subject of debate since its description by Tileston in 1906 as peptic ulceration of the oesophagus. Barrett himself initially theorised that the abnormal oesophagus was in fact stomach that had been pulled into the chest by a congenitally short oesophagus (1). This idea was ultimately challenged as the area in question lacked a peritoneal covering, contained submucosal glands and muscularis propria characteristic of the oesophagus (2). In 1976, Paull et al described a distinctive type of intestinal metaplasia the investigators called "specialised columnar epithelium”. Specialised intestinal metaplasia is now widely accepted to be the hallmark of BO with its presence predisposing to dysplasia and cancer regardless of its location within the oesophagus (3).
Barrett’s Oesophagus, its identification and treatment continues to be an area of debate and interest. Although not sinister in itself, it is a known precursor to malignant disease and strongly associated with GORD. Barrett’s oesophagus is the most frequent predisposing risk factor for the progression to adenocarcinoma in the oesophagus. Sufferers have a 40 fold increased risk when compared to the general population (4).
The progression of GORD to BO appears to be related to exposure of oesophageal tissue to the acidic contents of the stomach. It is therefore seen in hiatus hernia, lower oesophageal dysfunction, delayed oesophageal acid clearance and duodenogastric reflux. Furthermore, it is the duration and not frequency of exposure to acidity that dictates erosive damage to the oesophagus. Levels of acidity also contribute. The damage to cells incurred leads to inflammatory infiltration and cell necrosis with replacement of oesophageal epithelium by metaplastic columnar cells.
Assessing severity of BO relies partly on endoscopic visualisation techniques and length of oesophagus involved. Long segment Barrett’s oesophagus (LSBO) indicated a >3cm segment of involvement with short segment disease involving <3cm. LSBO carries a higher risk of progression to adenocarcinoma. Its development is associated with long term symptoms, severe combined patterns of reflux (both erect and supine) on 24 hour pH monitoring and reduced lower oesophageal sphincter pressures. Patients are less sensitive to direct acid exposure than those with short segment disease. The latter group also tend to have shorter duration of symptoms, normal sphincter pressures and only upright reflux on 24 hour pH monitoring (3).
The Prague C and M criteria is a recently developed classification system utilising the circumferential and maximal extent of oesophageal columnar tissue to assess disease severity endoscopically (5). Its accuracy is yet to be assessed clinically, however, it is believed to largely improve the overall assessment of Barrett’s (6). The further classification of disease severity is based on the degree of dysplasia, with high grade dysplasia carrying a higher risk of progression to malignancy.
Barrett’s oesophagus is predominantly seen in the age group 55-65, with males being affected twice as frequently as females. The disease is more prevalent in the white population. Obesity, smoking and alcohol intake being further risk factors. H.pylori may be protective against Barrett’s oesophagus with two mechanisms postulated. Namely, the induction of atrophic gastritis, which results in decreased acid production and the production of neutralising ammonia independent of gastric atrophy (7). The duration of symptoms of GORD but not necessarily symptom severity is also associated with increased risk of progression to BO. The exact pathogenesis is not clearly understood and is believed to be a culmination of both hereditary and environmental factors. For example, some studies report a greater incidence of BO amongst first degree relatives in comparison to their unrelated counterparts (8). Other reports associate environmental factors such as a high body mass index, with an increasing risk of GORD and progression to BO (9). Underlying mechanisms include the proposition that central obesity predisposes to hiatus hernia formation (10) and subsequent gastric acid reflux. However further research is required to unlock the key processes that lead to the formation of BO; as these pathways may hold novel therapeutic targets.
Prevalence of BO is difficult to ascertain due to the lack of population based studies. Studies from the United States involving patients aged over 40 years undergoing gastroscopy reported a prevalence of 6.8% in all patients (11). A Swedish study involving 1000 volunteers is the only available true population based study and found a prevalence of 1.6% (12).
Endoscopic surveillance
The most appropriate method for both diagnosis and surveillance of Barrett’s Oesophagus is endoscopy. Its sensitivity is higher than other comparative techniques, such as barium based studies or CT/MRI. Endoscopic screening programmes can be beneficial in both highlighting patients with BO from those with chronic GORD, as well as monitoring patients with established disease who are at risk of progressing to adenocarcinoma of the oesophagus. The American College of Gastroenterologists identify older patients with chronic GORD symptoms as the most likely to benefit from endoscopic surveillance techniques. Studies have also shown that five year survival rates are generally greater for patients who have had their adenocarcinoma identified by surveillance in comparison to those who have not (13). Importance also lies in the method of surveillance, for example shorter endoscopic interval analysis for surveillance in low grade dysplasia, are associated with higher rates of detection of adeonacarcinoma (14).
Although screening for Barrett’s oesophagus relies largely on established endoscopic techniques, it remains an area of contention for several reasons. These include low prevalence and the invasiveness of endoscopy, as well as a lack of an easily identifiable demographic group. Alternative methods include the use of capsule endoscopy which offers increased acceptability of screening, is less invasive and carries an increased uptake rate in comparison (15). However a study involving 96 patients demonstrated only 67% sensitivity and 84% specificity for identifying the condition using this technique (16). A recent meta-analysis of nine studies comprising 618 patients offers the most up to date evaluation of this technique. The pooled sensitivity and specificity for diagnosing BO using this method was found to be 77% and 86% respectively. Studies using OGD as reference demonstrated sensitivity 78% and specificity 90%. With intestinal metaplasia as the reference standard, sensitivity 78% and specificity 73% was discovered although the latter figure was particularly affected by one study with very low values for this (17). Capsule endoscopy offers benefits in patient tolerance and morbidity as well as cost as the capsule can be swallowed in an office, potentially under nursing supervision. Despite this latter point, cost-benefit analysis of this technique have proved equivocal. There are also several drawbacks. Views achieved are no longer under operator control and anatomical landmarks are more difficult and potentially impossible to identify. Oesophageal transition time has been demonstrated to be as short as 1 second and biopsy is not possible regardless of this. This greatly limits the use of capsule endoscopy in BO surveillance which relies on biopsy. Ultimately, the use of capsule endoscopy in diagnosis or screening of BO is unsupported at this current time and is an area for future research.
Other methods include small calibre trans-nasal endoscopy, which involves inserting a small-calibre endoscope through the nose and oesophageal sphincter to visualise the oespophagus, stomach and duodenum. It has the advantage of not requiring any sedation only topical anesthesia, having a lower complication rate, requiring less nursing staff and being more cost effective in comparison to its more frequently performed counterpart. Capsule endoscopy, as described earlier, also has the advantage of lacking sedation, being less invasive and yielding lower complication rates. Other alternatives include narrow band imaging, which involves scanning large areas of mucosa for possible neoplasia and autoflourescence imaging in which dysplastic lesions are visualised by differences in colour. The usefulness of visualisation techniques including high-resolution magnification endoscopy and tissue staining with agents such as methylene blue or indigo carmine are still an area of debate. These techniques have been evaluated when used in combination and alone. Pit patterns identified using acetic acid chemoendoscopy were described in 2001 by Guelrud et al (18) and Sharma et al described differing mucosal patterns in BO (19). Numerous other agents and classification systems have been described. Currently the use of these techniques for diagnostic purposes has not been shown to offer superior results than the current gold standard of four quadrant biopsies. Comparison of biopsies taken with methylene blue directed biopsy versus conventional biopsy showed no significant benefit (20). The ability to identify areas of BO (particularly high-grade dysplasia) are not in question. However, low grade dysplasia may be missed and operator experience and skill must be greatly superior to utilise the benefits of these techniques. Staining techniques offer the additional complications and additional expense of carrying out the procedure. Methylene blue has been shown to induce cellular DNA damage in vitro via the generation of singlet oxygen when photoexcited by light (21) thereby potentially being carcinogenic in itself. Evidence to support non-biopsy detection of BO is currently not sufficient to replace the current gold standard but is another area of current and future research.
The low prevalence of BO in the general population makes screening, with upper GI endoscopy, less viable on both a financial and logistic level. The general consensus is those individuals who suffer from chronic GORD are most susceptible to BO and would therefore benefit the most from upper GI endoscopy (22). However the factors involved in the progression of BO to dysplasia and subsequent adenocarcinoma remain unclear, and hence the value of endoscopic surveillance remains a point of discussion.
Treatment options
The treatment options for BO must also be taken into consideration when addressing surveillance and burden of the disease. The treatment options can broadly be divided into three groups, which include conservative management with surveillance endoscopy, endoscopic therapy and surgical oesophagectomy. The pathways of treatment are governed by patient-specific factors as well as the degree of oesophageal dysplasia. Surveillance endoscopy forms an integral part of the management of BO, and this is largely due to studies which have demonstrated a greater five year survival and an earlier stage of detection of oesophageal carcinomas detected by surveillance endoscopy (13, 23). Current recommendations target individuals at high risk of BO, for example those with chronic GORD symptoms. If no dysplasia is found on biopsies from two endoscopies, surveillance intervals of 3 years are recommended. However, patients with low-grade dysplasia on biopsy should have an immediate repeat endocopy to confirm the diagnosis, and then yearly surveillance endoscopies until no dysplasia is observed. The management of patients with high-grade dysplasia is contentious and varies between centres. Recommendations include a repeat endoscopy to evaluate for cancerous progression, with some centres instituting regular three month surveillance with biopsies every 1-2cm of effected mucosa (6). Other centres, depending on the multi-focal extent of dysplasia recommend surgical intervention with oesophagectomy or endoscopic therapy; which includes mucosal resection, photodynamic therapy, argon plasma coagulation and endoscopic ablative techniques.
Surgery
Oesophagectomy is normally reserved for the management of high grade dysplasia with the potential for malignant transformation. The percentage of high grade dysplasia which progress to adenocarcinoma vary throughout the literature from 5% to 59% up to seven years from initial diagnosis (7). Although oesophagectomy provides potential for complete resolution, it also carries increased number of adverse effects which include strictures, infections and anastomotic leaks. Mortality rates may also exceed 18% in centres which perform smaller amounts of the procedure on average every year (24) in comparison to high volume centres where the mortality rates can be lower than 5% (25); making the procedure very operator-dependent. As a result less invasive therapeutic modalities are preferred in the management of lower grade oesophageal dysplasia.
Endoscopic Therapy
Endoscopic treatment of Barrett’s oesophagus is currently an area of great interest. Endoscopic resection alone, or in combination with other treatments, have been investigated thoroughly in the past; however studies including large populations based and long term standardised protocol are lacking. The interpretation of these results is therefore very difficult.
Endoscopic mucosal resection (EMR) for high grade dysplasia in BO was first reported in 2000 (26). The procedure involved initial identification of macroscopically visible or chemoendoscopically identifiable Barrett’s lesions. If the lesion showed no evidence of penetration into deeper tissue or metastasis, confirmed by ultrasound guidance, it would be open to resection (27). Ideal lesions include those easily identifiable by macroscopic techniques, limited in size and restricted to the mucosa. However, almost all reports realised the risk of incomplete treatment with recurrence of disease. Some authors advocated the use of circumferential endoscopic resection in order to minimise this risk (28). Endoscopic ultrasound has also been used, prior to treatment, to optimise therapy and has a degree of use in staging of oesophageal cancers (29). Post EMR data showed a low rate of complications with high rate of complete eradication of Barrett’s tissue in the short term. Larghi et al (30) investigated the long-term follow-up of patients undergoing EMR and complete Barrett’s eradication (CBE-EMR). This study involved 24 patients over a 3 year period. Histological eradication of Barrett’s oesophagus was achieved in 87.5% of patients. 3 patients suffered strictures which were endoscopically resolved. Other studies have shown similarly successful eradication with similar complications of bleeding and stricture formation (31, 32). Comparison with previous studies also demonstrated the need for long follow-up to identify potential disease recurrence. In order to minimise stricture formation, a maximum cirmcumference of 50% could be resected during each therapy. A median of 2 sessions was required for complete eradication. 13 patients also received argon plasma photocoagulation in order to ablate isolated islands of a few centimetres of BO. These studies highlight the use of mucosal resection either alone or in conjunction with other treatment modalities, such as argon plasma coagulation, in the treatment of BO. Other options are discussed below.
Argon Plasma Coagulation
This procedure involves the use of a high voltage current to ionise a jet of argon gas and treat the effected tissue. It is also used to treat bleeding lesions endoscopically hence the term coagulation. This procedure has been suggested to be of use in the treatment of BO (33, 34) and several studies have evaluated its efficacy (35-37). Conclusions have been mixed with some studies showing high rates of Barrett’s recurrence and others also suggesting poor rates of initial lesion ablation (37). Generally, rates of complete reversal of BO range in the region of 61-70% (38-42). Other studies have shown more successful results with complete ablation in 87-100% of patients (43-46). A later study evaluated these results as well as performing a further long-term follow-up of 66 patients with high-grade dysplasia undergoing APC with anti-reflux treatment. Histologically confirmed Barrett’s oesophagus was found in 12.1% of patients during further endoscopic surveillance. Patients were treated with anti-reflux therapy (both medical and surgical) and one repeat session of APC. No intraepithelial neoplasia or oesophageal adenocarcinoma was detected during the entire follow-up period of 51 months median (range 9-85) (35).
The available evidence in relation to APC still remains slightly difficult to interpret. Even the larger trials do not involve extensive samples of patients. Furthermore there is a variance between studies with regard to patient selection and exclusion criteria, anti-reflux strategies and the procedure itself. Other pitfalls include the difficulty in assessing the precise depth of the lesion and whether the penetration during treatment was successful enough to ablate the entire lesion. There is also no histological confirmation to help correct insufficient ablation and for this reason some studies have reported an increased risk of progression to cancer and metastasis if invasion past the muscularis occurs (47). Low rates of recurrence seem to be related to the use of higher power settings for ablation, up to 90W as demonstrated by Madisch et al (35). This group also demonstrated the potential role for high dose proton pump inhibitor therapy using a total of 120mg daily in three divided doses to suppress acid for the duration of treatment. Surgical anti-reflux procedures were also found to be associated with reduced recurrence rates. As mentioned above, this procedure may in itself provide a form of treatment for BO and further progression.
Although rare, complications of APC can be severe. Oesophageal perforation has been reported with 2 patient deaths as a consequence (40, 42). Mild oesophageal strictures amenable to endoscopic dilatation have been widely reported. Pleural effusions and bleeding ulcers have also been reported. Despite this, APC can be useful as an adjunct and also effective in the treatment of distinct groups of Barrett’s sufferers with amenable lesions.
Photodynamic therapy
Photodynamic therapy (PDT) involves the systemic administration of a photosensitising drug, followed by irradiation with a controlled light source via an endoscope. The light, in the presence of oxygen, activates the photosensitiser causing photochemically induced tissue destruction (48). Although technically this sounds a difficult procedure, in practice it is actually one of the simplest to perform. However, as with surgical oesophagectomy, it is operator dependent with complication rates increasing within the community in comparison to specialist centres.
The component parts of the photosensitisation process have also evolved with time.
Several photosensitisers have undergone trial with varying results. Trials with Hematoporphyrin derivative as the photosensitising agent showed high rates of stenosis as well as prolonged sensitivity of the skin to light (49). More recently, 5-aminolevulinic acid (5-ALA) has shown promise with good therapeutic results and reduced side-effects in the short term (50-51). 5-ALA also has a reduced period of cutaneous photosensitivity of around one week, in comparison to previous photosensitisers such as sodium porfimer, in which patients would need to take precautions for thirty to ninety days (27). Several studies have demonstrated the effectiveness of photodynamic therapy in BO. The first randomised clinical trial looked at 485 patients with BO and high grade dysplasia (HGD) (52). 208 patients were accepted into the to-treat population, and received photodynamic therapy and omeprazole (PDT+OM); whilst 202 patients formed the control group and received omeprazole alone (OM). The study demonstrated a significant difference with 77% of the PDT+OM group, compared with 39% of the OM group, receiving complete ablation of HGD. The progression from HGD to adenocarcinoma was also significantly lower in the PDT+OM group (13% vs 28%). This study highlighted the effectiveness of photodynamic therapy in conjunction with medical antacid therapy, in ablating high grade dysplasia and reducing the incidence of oesophageal adenocarinoma.
Pitfalls of PDT include the suggestion that lesions greater than 2mm in depth cannot be effectively removed (53), although the photosensitising agent used can influence this. For example, some studies have shown sodium porfimer to have an increased treatment depth of 3-4mm in comparison to other agents (54-55). However limited depth of penetration overall can compromise the ability of PDT to effectively treat high-grade dysplasia. Other common complications post PDT include stricture formation with some studies reporting rates as high as 30% overall and 50% in patients undergoing more than one procedure (56). Although high, long term complications related to this are not reported and most cases are relieved with endoscopic dilatation. Similarly other endoscopic techniques, photodynamic therapy may be inadequate at eliminating dysplastic tissue that is not visible on endoscopy. The issue of buried glands is an area of great interest due to the implication that a treated patient with macroscopically normal tissue may have dysplastic or even malignant tissue beneath. This highlights the importance of regular follow-up endoscopies with a thorough biopsy protocol. An additional complication with photodynamic therapy is the lack of histological samples post therapy, which might be used to assess the completeness of resection as in EMR.
Despite its limitations, photodynamic therapy has been proven an effective treatment for BO in numerous trials and case reports. Future directions include steps to improve photosensitiser agents, dosimetry, and light parameters which should help minimise the associated complication rate.
Radiofrequency Ablation
Radiofrequency ablation is one of the newer endoscopic treatment modalities to show promise in preventing the progression of Barrett’s oesophagus and eliminating the lesion completely. The technique utilises a balloon, 3cm long and consisting of a 60 electrode rings spaced narrowly together every 500micrometres in a bipolar fashion (HALO360 system, Barrx Co, Sunnyvale, CA, USA). A sizing balloon is used to ascertain the circumference of the area to be treated before the ablation balloon is introduced. The system then delivers radiofrequency energy to the tissue circumferentially for 300milliseconds. A dose of 12J/cm2 has been shown to be effective in achieving depth penetration accurately above the muscularis mucosae thus limiting the complications involved with damaging deeper tissues (57). The close spacing of electrodes allows uniform penetration of the entire treated circumference and thus this technique can be used circumferentially with reports of stricture formation being minimal (58). This ability to control the depth of ablative penetration means that many other adverse side effects seen with alternative endoscopic techniques are greatly reduced. These include lower rates of chest pain, odynophagia, perforation and pneumothorax in comparison with laser and thermal ablation techniques.
One recent paper reviewed the progress of 142 patients with endoscopically identifiable Barrett’s oesophagus and high-grade dysplasia managed at 16 separate academic and community centres. These patients underwent a total of 229 radiofrequency ablations and were followed up with repeat endoscopy and systematic biopsy for a median length of 12 months. The only adverse event of note was a stricture noticed on endoscopy in an asymptomatic patient. At follow-up, biopsy specimens were negative for high-grade dysplasia in 90% of patients. 80% of patients had no dysplasia on biopsy and 54% of patients were negative for intestinal metaplasia (59). These results are very encouraging, particularly as high-grade dysplasia carries the greatest risk of malignant progression.
Other benefits include minimal post-procedure discomfort with patients able to go home within hours of the procedure. Regarding the issue of buried glands, a study following 102 patients post circumferential ablation showed no evidence of buried glands in 4306 biopsy samples taken over a year follow-up (60). This once again highlights the advantages of RFA in comparison to other endoscopic techniques.
Conclusion
The surveillance and treatment of Barrett’s Oesphagus remains an area of interest and controversy. This is heightened by the inability to discriminate those patients with BO which are most likely to progress to high grade dysplasia and then to adenocarcinoma of the oesophagus. This places greater emphasis on the endoscopic surveillance programme to identify this potentially pre-malignant state at an early stage. Future advances, particularly in endoscopic techniques, will help to increase efficacy of treatment and minimise complication rates. Further developments include progress in identification of genetic biomarkers which may help elucidate those patients at greatest risk. The management of Barrrett’s Oesophagus is becoming increasingly more important, particularly with the rise in incidence of oesophageal carcinomas in the Western world. The issues to address therefore include the identification and screening of at-risk groups and the further management from diagnosis of BO. Patients with chronic GORD symptoms are most in need of screening. Currently this should include the gold-standard four quadrant biopsy technique. This may include techniques to enhance visualisation as described above. In the authors opinion, non-biopsy screening does not carry enough evidence to support its use in replacement of biopsy as of yet. Medical treatment with PPI (if necessary in high-dose) as well as surgical treatment of GORD are essential considerations in the prevention and treatment of BO. Their use in combination with endoscopic therapy has proven benefits as outlined. Of the endoscopic therapies, the lack of complications combined with excellent post-procedure rates of disease elimination seen with RFA are most encouraging. Oesophagectomy should be reserved for those patients with disease not amenable to conservative or endoscopic therapy. Continual research is required to help us gain more understanding into the pathogenesis of this condition, enabling us to effectively target and manage BO appropriately.
According to The National Kidney Foundation of The United States of America 1 CKD is defined as (1) evidence of kidney damage based on abnormal urinalysis results (e.g., proteinuria, hematuria) or structural abnormalities observed on ultrasound images or (2) an absolute GFR of less than 60 mL/min for 3 or more months. Based on this definition there are five stages. See Table 1. Anemia affects 60% to 80% of patients with chronic kidney disease (CKD) and reduces their quality of life. Treatment options are blood transfusion, epoietin alfa and darbepoetin alfa 2 . Anemia of CKD is, in most patients, normocytic and normochromic and primarily caused by depressed production of erythropoietin (EPO), oxidative stress and inflammation, erythropoiesis inhibition and reduction in red blood cell survival 3,4,5 . The other cause of anemia is deficiency of iron. The dialysis patient is in a state of continuous iron loss from gastrointestinal bleeding, blood drawing, and/or, most important with hemodialysis (HD), the dialysis treatment itself. HD patients lose an average of 2 g of iron per year. Thus, iron deficiency will develop in virtually all dialysis patients receiving EPO unless supplemental iron therapy is given orally or intravenously. DRIVE study, a randomized trial study, adds direct evidence that administration of intravenous iron to patients with functional iron deficiency who were on supplemental EPO therapy results in increase in the hemoglobin level 6. The aim of this review is to assess that whether Erythropoietin (EPO) treatment is beneficial or harmful in the management of anemia associated with CKD. To address these issues, we have analyzed randomized controlled trials, observational studies and meta-analyses.
Table 1: Stages of CKD according to NKF.
CKD Stage
Kidney damage
GFR
Stage 1
No kidney damage
>90 mL/min
Stage 2
mild kidney damage
60-90 mL/min
Stage 3
moderate kidney damage
30-59 mL/min
Stage 4
severe kidney damage
15-29 mL/min
Stage 5
Endstage kidney damage
<15 mL/mi
Methods:
Search strategy: The search strategy was designed to capture the patient population, suffering from CKD on supplemental erythropoietin (EPO) therapy. Literature search (1989 to 2008) was carried out using MEDLINE, PubMed as well as other electronic databases and in online journals using the keywords "Kidney failure, chronic and "erythropoietin" for studies up to 1996, and "epoetin alfa" for subsequent years.
Selection of studies: All papers identified were English-language, full text papers. In addition, the reference lists of identified relevant articles were also searched. The search was not limited to any specific study design, and we searched for randomized controlled trials (RCTs), observational studies, systemic review and meta-analysis. Citations identified in the literature search were independently screened by author to select potentially relevant articles. The full articles from this list were retrieved and subsequently reviewed by author for inclusion in the systematic review. During selection preference was given to articles published within last five years. Articles were included if they met the following inclusion criteria 1) published in a peer reviewed journal; 2) written in English; 3) reported randomized controlled trials of EPO; 4) observational studies regarding EPO and quality of life; 5) Review articles and meta-analyses about EPO therapy in CKD patients.
Results:
470 citations were identified in search from PubMed, Medline and from other online journals. Of these 470 citations, 444 did not meet the selection criteria and were excluded, leaving 26. Out of 26 citations 11 were RCTs, 10 were observational studies and 5 were reviews and meta-analysis.
Five studies (Parfrey et al7, Foley et al8, Furuland et al9, Drüeke et al10, and Canadian Erythropoietin Study Group11) showed that correction of anemia result in improvement of quality of life, although the singh et al12 showed such improvement with partial correction of anemia, and no detectable difference in the quality of life was evident in Roger et al13 study. Five studies Parfrey et al7, Foley et al8 and Levin et al14 and McMahon et al15 and Roger et al13 showed that normalization of hemoglobin does not lead to regression of established concentric LV hypertrophy or LV dilation. It may, however, prevent the development of LV dilation. In McMahon et al15 study the only factor that seemed to predict normalization of LV mass in patients who had LV hypertrophy at study entry was a lower pulse pressure. One study Sikole et al16 correction of renal anemia can normalize heart morphology and improve heart function. Three studies Besarab et al17, Drüeke et al10 and Singh et al12 demonstrated increased cardiovascular events whereas two studies Drüeke et al10 and Singh et al12 also showed progression to dialysis in patients assigned to the highest hemoglobin targets (>13.0 g/dL), compared with <12 g/dL, trial design of three studies was same in respect that both arms were on EPO. In comparison to Drüeke et al10 and Singh et al12 studies, in Roger et al13 study the renal function was not adversely affected in the group randomized to the higher Hb. (Table 2).
Table 2: Randomized controlled trials (RCTs)
Study
Study design
Patients enrolled
Parameters observed
Outcomes
Parfrey et al7
RCT double blind
596
QoL, LVVI
No change LVVI. QoL improved.
Foley et al8
RCT open label
146
QoL, LVH, LVD
QoL improved, No change LVMI
Furuland et al9
RCT open label
416
QoL, Safety.
QoL improved.
Drüeke et al10
RCT open label
603
CVE, QoL, LVMI, Renal function
QoL improved, No change LVMI.
Canadian Erythropoietin Study Group11
RCT double blind
118
QoL .
QoL improved.
Singh et al12
1432
CVE, QoL, Renal function
QoL not improved.
Roger et al et al13
RCT open label
155
LVMI. Renal function, QoL
No change.
Levin et al14
RCT open label
172
LVMI.
No change.
McMahon et al15
RCT open label
120
Change in LVMI.
Prevention&↓ in LVMI.
Sikole et al16
RCT open label
38
Heart morphology & functions.
Heart function
Improved.
Besarab et al17
RCT open label
1233
Effects of normal HCT.
↑CVE
Both Phrommintikul et al18andGiovanni et al19 addressed the similar issues, to evaluate the benefits and harms of different hemoglobin (Hb) targets in CKD in their meta-analyses. They reached to similar conclusion, increase in the risk of all-cause mortality in anemic patients with CKD in whom a higher Hb target (in the normal physiological range) is aimed for with treatment with EPO. Such patients are also at an increased risk of arteriovenous access thrombosis and poorly controlled hypertension, which could contribute to the increased risk of mortality. Furthermore, there seems to be no beneficial effect on left ventricular mass in such patients. There were similarities and differences in inclusion criteria. Both used the trials targeting different Hb concentrations in patients with anemia caused by CKD, majority of trials analyzed were different except 4 trials which were same in both. The difference in inclusion criteria was that Giovanni et. al analyzed two groups of studies: The first group contained studies in which the intervention was to achieve different Hb targets compared (higher versus lower Hb targets), both arms were on EPO and trials included individuals with clinical cardiovascular disease. The second group compared EPO treatment with no EPO treatment. The results of these two groups of studies were analyzed separately. In Phrommintikul only one group of studies “EPO treatment with no EPO treatment” was analyzed. Meta-analysis by Phrommintikul et alincludes nine RCTs, which enrolled 5143 patients.
In Jones et al20 meta-analysis both randomized controlled trials and uncontrolled trial were analyzed, all studies were of the “pre/post” design, in that measurements of anemia, quality of life, hospitalizations, and transfusions were taken before and after initiation of EPO therapy. They drew these conclusions from 16 published studies of which 5 were randomized clinical trials. He found that treatment with EPO raised hemoglobin levels, reduced transfusion requirements and improved quality of life. For quality of life outcome in these meta-analysis please review Table 3.
Table: 3 Meta-analysis.
Meta-analysis
Question addressed
No of studies included.
Assessed quality of life (QoL).
What measures of quality of life used in these trials.
Were the major gains in QoL seen with EPO.
Phrommintikul et.al18
Target Hb and cardiovascular events in CKD.
9
No
_____
_____
Giovanni et al19
Evaluate the benefits and harms of different Hb targets in CKD.
19
Yes
KDQ
QoL
Improved
↑KDQ
Jones et al20
Effects of
EPO on clinical efficacy, QoL hospitalizations,
and transfusions.
16
Yes
KPS
KDQ
SIP
QoL improved
↑KDQ
↑ KPS
↓SIP
Discussion:
Erythropoietin (EPO) has become an essential part of the management of anemic patients with CKD. It is also used to treat the anemia associated with chemotherapy and other diseases, and it improves quality of life 21,22 . The introduction of EPO in 1989 significantly improved the clinical management of anemia of CKD. By 2005, 99% of incenter hemodialysis patients received EPO treatment for their anemia. EPO dosing has changed dramatically in the past decade and a half; between 1991 and 2005, the mean dose of EPO increased about 4-fold in dialysis patients. Today, EPO therapy is the largest single Medicare drug expenditure totaling $1.8 billion in 2004 (an increase of 17% from 2003) and EPO comprised 11% of all Medicare ESRD costs 23
EPO and left ventricular hypertrophy. Randomized vs. Observational studies. Anemia is a contributing factor in many of the symptoms associated with reduced kidney function. These include fatigue, depression, reduced exercise tolerance, dyspnea.Data from observational studies shows that severe anemia may results in cardiovascular consequences, such as left ventricular hypertrophy (LVH) and left ventricular systolic dysfunction 24. Left ventricular hypertrophy (LVH) is present in nearly 80% of dialysis patients and is associated with higher rates of cardiovascular events 25. It is also associated with an increased risk of morbidity and mortality principally due to cardiac disease and stroke 26,27. As a result, patients with anemia due to CKD are at increased risk of hospitalization, hospital length of stay, reduced quality of life and mortality 28. Uncontrolled studies suggested that partial correction of anemia with EPO therapy may result in prevention or regression of CHF 29 and LVH 30,31. Several randomized controlled trials showed that left ventricular hypertrophy was not further improved by a complete correction of anemia compared to only partial correction 7,13,14,15,17. Robert N Foley et al. randomly assigned 146 patients with either concentric LV hypertrophy or LV dilation to receive EPO to achieve hemoglobin levels of 10 or 13.5 g/dL. He concluded that normalization of hemoglobin does not lead to regression of established concentric LV hypertrophy or LV dilation. It may, however, prevent the development of LV dilation, and it leads to improved quality of life 8. Partial correction of severe anemia <8 or 9g/dl to mild anemia (10-11 g/dL) likely reduces mortality, but further treatment to higher Hb levels has not been shown to further reduce mortality, and has actually increased mortality. Controlled studies with quality of life (QOL) and left ventricular mass as end points support partial correction of hemoglobin in dialysis patients 11,13,16,32, The fact that anemic renal failure patients have more LVH than non-anemic renal failure patients does not prove that anemia causes LVH. Vaziri et al in a review mentioned that the real culprits are oxidative stress, inflammation and diminished biological capacity that simultaneously cause treatment-resistant anemia and adverse cardiovascular and other outcomes 33.
EPO and quality of life:Numerous randomized, controlled trials have demonstrated that EPO significantly raises hemoglobin levels, reduces transfusion requirements, and improves quality of life in anemic patients with chronic renal failure. Lefebvre et al conducted an analysis on data from a multicenter, open-label, prospective study of EPO for anemia in patients with CKD not on dialysis to evaluate the relationship between Hb level and quality of life (QOL). The results showed that the maximal incremental gain in QOL occurred when Hb reached 11 to 12 g/dL. This suggests that treating anemic patients with non-dialysis CKD until their Hb level reaches 12 g/dL will result in the greatest QOL improvement per Hb unit increase 34.
Randomized vs. Observational studies. Many randomized controlled trials suggest an association between higher hemoglobin level and improved quality of life, physical function, and exercise capacity 7,8,9,35 but the association with survival is less clear. Whereas observational studies have generally shown increased survival with higher hemoglobin level 36,37,38,39,40 randomized trials have not shown such benefits. The extent anemia of inflammation varies between patients with renal failure. It is this factor that likely explains the following paradox: in observational studies, higher hemoglobin associates with better survival in CKD, while in controlled trials, higher hemoglobin achieved by escalated EPO dosing decreases survival. In the observational studies, those with the higher Hb levels were likely those patients who had the least component of anemia of inflammation, and therefore less resistant to EPO supplementation; they survived better not because they had better hemoglobin, but because they had less burden of inflammatory disease. Anemia of chronic disease is a highly conserved response that is mediated by multiple mechanisms acting in concert to lower the hemoglobin in the face of inflammation, and should be presumed until proven otherwise to be adaptive for most patients who exhibit it. That this is so is supported by the observation that the correction of anemia confers lower survival not only in renal failure, but also in cancer patients and in patients in the critical care unit.
Jones et al. in their very thorough meta-analysis 20 indeed found that treatment with EPO raised hemoglobin levels, reduced transfusion requirements and improved quality of life. Studies have demonstrated that morbidity and mortality rates are lower when hematocrit values are within the Disease Outcomes Quality Initiative (DOQI) target range (33 to 36%) 40. Ernesto Paoletti et alin their review of observational and randomized studies concluded from the results of observational studies that normalization of Hb in renal patients seems to be associated with further improvement in quality of life and physical activity but with no significant differences in mortality rate, hospitalization rate, and the extent of LVH regression, but the results of randomized trials show that achieving near-normal Hb did not reduce the risk for death from all causes or the risk for cardiac death. The latter risk actually increased slightly, in the group of dialysis patients with normalized Hb concentration 41. For CKD stage 3 and 4 patients, no improvement seen in CHOIR, but improvements reported in CREATE. There may be reporting bias in CREATE as it was an open label study, and the low target arm had to develop worsening anemia prior to initiating EPO therapy 10,12
EPO and Cardiovascular Events:Anemia is a common complication of chronic kidney disease. Determination of the appropriate target hematocrit level for patients undergoing hemodialysis continues to be a controversial area 40. The National Kidney Foundation Dialysis Outcomes Quality Initiative (K/DOQI) states when a decision to use EPO is made, some Hgb value in the range of 11 to 12, but no higher than 13 should generally be chosen. 42. The European Best Practice Guidelines (EBPGs) recommend that most patients with CKD achieve a target hemoglobin (Hb) 11 g/dl to reduce the risk of adverse outcomes 43.Randomized vs. Observational studies. Observational studies have shown a strong association between severity of anemia and risk of morbidity and mortality from cardiovascular disease and other causes in CKD patients 36,37,38,39,40. These findings have been interpreted as evidence for the causal role of anemia in the pathogenesis of adverse outcomes in these patients. On the hand, randomized clinical trials of anemia management revealed either no effect or increased morbidity and mortality in patients assigned to normal hemoglobin Hb targets 10,12,17. Meta- analyses of randomized clinical trials have shown a significant increase in cardiovascular and all-cause mortality and arteriovenous access thrombosis among patients assigned to the higher than those randomized to the lower Hb targets 18,19. Meta-analysis of Phrommintikul shows a significantly higher risk of all-cause mortality (targeting a Hb level higher than 12 g/dL results in a 17% increased risk of death compared with target hemoglobin levels less than 12 g/dL), arteriovenous access thrombosis and higher risk of poorly controlled blood pressure in the higher Hb target group than in the lower target Hb. The incidence of myocardial infarction was much the same in the two groups 18. Meta-analysis of Giovanni F.M et al shows that on the basis of available randomized, controlled trials, Hb targets of <12.0 g/dL are associated with a lower risk of death in the population with cardiovascular disease and CKD compared with Hb targets of >13.0 g/dL. For every 30 patients treated to an Hb target of <12.0 g/dL compared with an Hb target of >13.0 g/dL, approximately one death is avoided 19. Two large randomized controlled trials; CREATE [38] and CHOIR [39] demonstrated increased cardiovascular events and progression to dialysis in patients assigned to the highest hemoglobin targets (>13.0 g/dL), compared with <12 g/dL. US Food and Drug Administration (FDA) warned that use of erythropoiesis-stimulating agents (ESAs) increases mortality and morbidity risk. The warning follows publication of studies suggesting that correction of anemia in patients with CKD did not reduce the risk of cardiovascular events and that reaching a target Hb level of >13 g/dL, compared with a target level of 11.3 g/dL, was associated with increased risk of cardiovascular events. FDA said recent studies had found increased risk of death, blood clots, strokes, and myocardial infarctions in patients with chronic renal disease who received ESAs at higher-than-recommended doses that maintained their hemoglobin levels at more than 12 g/dL 44.
EPO and kidney:EPO has been found to interact with its receptor in a large variety of non-haematopoietic tissues, which result into cytoprotective cellular responses, like mitogenesis, angiogenesis, inhibition of apoptosis and promotion of vascular repair through mobilization of endothelial progenitor cells from the bone marrow. In experimental ischaemic and toxic acute renal failure administration of EPO, promotes renoprotection. Preliminary experimental and clinical evidence also indicates that EPO may be renoprotective in chronic kidney disease22 .EPO is used widely to treat anemia in patients with CKD, but the benefits of EPO use in patients with acute renal failure (ARF) are unclear. In vitro and animal studies suggest that EPO may promote renal recovery and decrease mortality in ARF 45. Partial amelioration of anemia with low doses of EPO was reported to slow the rate of progression to ESRD in a group of CKD patients 46. These cellular protection from EPO observed in animal models has not been confirmed in humans, and has been specifically addressed and disproven in large randomized trial. The CREATE study found as a secondary endpoint that early treatment with EPO increased the likelihood of starting dialysis. CHOIR found no reduction in the rate of progression of CKD in patients given more EPO (the higher target arm) compared to the lower target arm 10,12.
EPO for other Indications:In recent years, studies in animals and humans have focused on the use of EPO for other indications. It has been found to play a role in both cardioprotection and neuroprotection. It has effects on the immune system, and can cause regression in hematologic diseases such as multiple myeloma. It may also improve the response of solid tumors to chemotherapy and radiation therapy 21. Again the cellular protection from EPO observed in animal models has not been confirmed in humans.
EPO and Seizures:Seizures are reported to be a complication of EPO in the product information. However, in a meta-analysis conducted by Giovanni et alshowed that treating with EPO may be protective against seizures Lower Hb targets of <95 g/L in individuals who are not treated with EPO are associated with a significantly increased risk of seizures compared with treatment with EPO and Hb values of >100 g/L 19.
EPO and Hypertension:Administration of ESA may be associated with exacerbation of
hypertension in about 5% of patients. Robert N. Foley et alin his analysis of observational and randomized studies found that most of the trials that have been reported to date have shown that higher hemoglobin targets lead to higher BP levels and/or greater requirements for antihypertensive therapy, he drew this conclusion from nine randomized trials 47. The mechanism of ESA induced hypertension is thought to be related to stimulation of the vascular endothelium by ESA resulting in increased circulating levels of endothelin. Furthermore the increase in hemoglobin associated with ESA therapy may increase blood viscosity resulting in vasospasm. As such routine monitoring of blood pressure is essential in patients treated with ESA 48. Meta-analysis of Phrommintikul et al showed a significantly higher risk of poorly controlled blood pressure in the higher haemoglobin target group than in the lower target hemoglobin 18 . Giovanni et almeta-analysis showed lower Hb levels of <95 g/L with no EPO treatment are associated with a reduced risk of patients who present with hypertensive episodes. In absolute terms, the risk of developing hypertensive episodes is 16% lower with Hb values <95 g/L compared with Hb >100 g/L. For every seven patients treated to obtain an Hb >100 g/L, one patient will require additional antihypertensive medication 19
EPO and Access:Normalizing hemoglobin has been associated with a higher incidence of vascular access clotting 40. Randomized, prospective, open-label trial study of Besarab et al, showed a significantly higher risk of access thrombosis with the higher Hb targe17. Meta-analysis of Phrommintikul et alshowed a significantly higher arteriovenous access thrombosis in the higher Hb target group than in the lower target Hb 18.
EPO and Pure Red Cell Aplasia (PRCA):Although rare, administration of ESA may result in formation of anti-erythropoietin antibodies, thereby leading to pure red cell aplasia and erythropoietin resistance 49 In patients in whom ESA doses have been maximized without effect and no other causes can be identified, serum anti-erythropoietin levels and bone marrow biopsy should be performed. If confirmed, erythropoietin administration should be ceased and the patient treated with periodic blood transfusions.
Conclusion: Achieving hemoglobin control over time is a major challenge because of the various physiological factors that influence the response in individual patients and the potential risk for increased mortality, particularly for patients with co morbidities 50. The association data led to several hypotheses about what anemia was causing e.g. LVH, fatigue, and increased mortality. These hypotheses have been tested in RCT's and in most cases anemia is associated with but does not cause the outcome, such as LVH or mortality. Fatigue is improved somewhat by anemia treatment with EPO, and transfusion frequency is reduced, though the cost is high. In the case of EPO balance is critical. Too little treatment and patients with chronic kidney disease are subjected to a lifetime of exhaustion and blood transfusions. Too much and they could be threatened with an increased risk of death. The overall quality of life is improved when anemia is treated with EPO, but aiming for a target value of 13.5 g of hemoglobin per deciliter provided no additional quality-of-life benefit 12.
Key Points:
Anemia is associated with bad outcomes; Anemia is nearly universal in advanced renal disease. In these patients, anemia is associated with increased cardiovascular morbidity and mortality, reduced quality of life, and accelerated renal disease progression, though those associations do not necessarily establish causation.
Treatment of anemia reduces transfusion requirements and improves quality of life in anemic patients with CKD.
Mortality increases with treatment to higher targets; Recent studies have found an increased risk of death, blood clots, strokes, and myocardial infarctions in patients with chronic renal disease who received ESAs at doses that maintained their hemoglobin levels at more than 12g/dL, leading the Food and Drug Administration to apply a 'black box' warning to the product monographs of licensed ESAs.
Recent studies support partial correction, not normalization of hemoglobin.
Good Medical Practice describes the professional behaviour expected of doctors and advocates that it should be taught as well as assessed. GMC’s Good Medical Practice specifies the standards of team working, communication skills, accessibility and trustworthiness in relation to professional behaviour1. Ramsden2 stated that: “the students will learn what they think they will be assessed on, not what is in the curriculum, or even on what has been 'covered in class'.” Hence, if the intended learning objectives are to improve professional behaviour and team working in a trainee doctor, then a tool that assesses these characteristics along with providing suggestions for improvement is critical. Multisource Feedback (MSF) is a formative assessment tool that was designed to assess professional behaviour and attitudes, with the aim of continually improving an individual’s team working. Team Assessment of Behaviour (TAB) is an assessment tool for MSF (Appendix 1), and is one of the 2 assessment tools used to assess professional behaviour of foundation doctors in training in the UK, the other being the Mini-Peer Assessment Tool (mini-PAT)3. Our aims for this article were to evaluate whether the format of TAB allowed the MSF process to occur as originally intended, along with looking at possible barriers that may have to be overcome to make it an effective tool. A literature review was carried to appraise the present evidence regarding TAB as an assessment tool and only studies which had relevance to MSF/TAB were included in the study. As TAB is a relatively new tool, there were very few papers’ exploring this is depth which was a major limitation. Hence in the barriers section, we have discussed the possible obstacles to the whole MSF process rather than just TAB. Background The concept of MSF was originally developed by industrial organisations and has been used in postgraduate medicine in USA for assessment of professional behaviour since the 1990’s4,5. Ramsey6 suggested that it is feasible to obtain assessments from professional associates of practicing physicians in areas such as clinical skills, humanistic qualities, and communication skills. TAB was developed by educationalists and senior doctors at the West Midlands deanery7,8,and has undergone extensive field testing among 171 trainees, with analysis of received feedback from 1378 assessments across four different hospitals in the West Midlands. It is currently being used in the West Midlands Deanery for the multisource feedback of foundation programme trainees. Process of TAB The primary aim of TAB as an assessment tool is to identify trainees whose professional behaviour does not meet GMC requirements for good medical practice, so that appropriate action may be taken and also to compliment those trainees who receive good reports. For TAB, up to 10-15 multi-disciplinary colleagues of a doctor assess his/her workplace behaviour. It assesses four domains of professional behaviour: professional relationship with patients, verbal communications, team-working and accessibility. It is the initiative of the trainee to distribute at least 15 TAB forms to peers of their own choice and a minimum of 10 completed forms are required to be returned. The raters should include at least three other doctors including a consultant supervisor and at least five allied healthcare professionals. It’s the responsibility of the educational supervisor to collate and summarise these forms, identifying perceived weaknesses, offering them feedback and directed learning objectives to address any issues. Critique of TAB:1. Validity and reliability of TAB:Validity is a demonstration that a particular instrument can in fact measure what it purports to measure9. TAB portrays face as well as content validity as it assesses areas identified by the GMC1 for good professional behaviour. It is shown to be capable of identifying problem behaviour in trainees, which was the primary aim of the tool7. This tool appears to have good construct validity as it is testing trainee’s behaviour in real life situations. It is difficult to define the predictive validity of any tool, more so in a formative assessment tool. The developers of TAB have not tested for concurrent validity. The concurrent validity for TAB could have been tested by using another MSF tool in addition to TAB for some participants during the field-testing for reliability and validity of the tool. A reliable instrument for a piece of research will yield similar data from similar respondents over a period of time9. Whitehouse7 demonstrated in the pilot study that TAB had intra-observer reliability and inter-observer variability. For inter-observer variability, the Royal College of Psychiatrists compiled a map of assessment programmes against good medical practice domains and considered it appropriate for assessing four domains: good clinical care, working with colleagues, probity, and health11. 2. RatersRamsey et al5 concluded that, with MSF, 10-11 responses per physician were necessary to achieve a generalisablity coefficient of 0.70. Wood et al11 concluded that eight raters were sufficient for a representative score in their study on Obstetrics & Gynaecology trainees in the UK. Obviously more raters would lead to better coefficient and more generalisable results. TAB presently advocates at least ten raters to achieve reliable results.3. Feasibility TAB has four domains and a three point rating scale which are relatively easy to understand and complete. There is no training required for raters and usually takes less than five minutes per assessment per assessor. The paper-based system demanded considerable administrative resources, and therefore a web-based TAB assessment form was successfully piloted in the West Midlands8.4. Trainer & trainee’s evaluation of the processThe evaluation of TAB as an assessment tool was done by Whitehouse et al8 as a part of their field assessment of TAB. The assessors and trainers found the process practical, valuable and fair. 76% of the trainees who responded to the questionnaire felt that it was a useful addition to the assessment of the SHO’s. The educational supervisors had mixed views, with 77% of them finding out nothing new about the trainees. 5. Scoring systemInstead of a Likert scale, the TAB employs broad boxes which offer the rater a choice of giving specific feedback under each domain. This is more helpful to a summariser/assessor than mini-PAT, where a scoring scale is not substantiated by relevant feedback. This assessment tool does not assess clinical performance and one could argue that there could have been more than four domains in order to include other areas of performance, such as clinical skills. However having just four domains can reduce the impact of the halo effect. The halo effect12 can be defined as a rater making an overall judgement of the trainee and scoring the whole form accordingly rather than considering each domain separately. This could be a potential advantage of TAB over mini-PAT where there is no opportunity to provide specific feedback in individual domains, but rather utilises an overall action plan, which may lead to concentrating single element deficiencies while masking other shortcomings. 6. Patient OutcomesThe MSF process in itself doesn’t bear direct consequences on patients’ management, but can help the doctor improve his professionalism, which is critical element of good medical practice1. An improvement in a doctor’s behaviour secondary to the appraisal-feedback process can indirectly improve and contribute to patient management and satisfaction. Potential barriers: 1.Choice of rater:The trainee has the choice of raters, except for the supervising consultant, who must be involved in the process. This is a potential area of difficulty, as the trainee might pick individuals who are more sympathetic to their cause, or who cannot comment much on their interpersonal behaviour, thus leading to skewed results. Kuzmits13 showed that both rater and those being rated needed to be trained to make the rating and feedback process more effective, but this is not felt necessary for TAB. There is also a potential conflict between being a trainer and assessor, and this conflict might lead to clouding of judgement14, which can be addressed by having different assessors and trainers, but considering the service pressures in the NHS, this may not always be possible. 2. Feedback:The value of the MSF process can be limited by the quality of feedback provided to the trainee at the end of the process and depends on the relationship of the trainee and the supervisor15. Evidence shows that non-specific feedback does little to change performance16. Whitehouse8 concluded in their initial study that TAB was able to produce descriptive feedback that was more specific and helpful than existing MSF tools. Assessors completing TAB may not give specific comments or feedback although they are instructed to give some details especially if they chose the rating of “some concern” or “major concern”. Bret and Atwater17 have shown that negative feedback can discourage individuals, and they can even react in anger. Feedback about performance must be descriptive and specific if it is to be helpful to trainees18. Hence it is the responsibility of the supervisor to give feedback which is relevant and helpful, along with creating an action plan with the trainee to address any perceived deficiencies. It must also be stressed that good performers need to be complimented, and encouraged to continue to do so. 3. Training the raters and the supervisor:The raters also need to be educated in the process otherwise they may not give reliable views about the trainee13,19. From the rater’s point of view, it would be beneficial if they were to give specific comment, in order that more relevant feedback can be provided. Most importantly, the educational supervisors who provide the feedback to the trainee can make a difference in constructing agreed learning objectives, and not demoralise the trainee with negative feedback. Kaplan20 also noted that negative feedback can demotivate individuals. Giving face to face negative feedback can be a daunting task and supervisors, if not trained in giving negative feedback may dislike doing it, and so not give properly constructed feedback. The provision of training, however, has time as well as cost implications. Conclusions TAB continues to be part of the national foundation programme curriculum and, used correctly, can serve its purposes both as a screening tool and also for the trainees to use the feedback provided to improve their interpersonal behaviour when needed. Evaluation and quality assurance of this assessment tool should be an ongoing process. More field work in relation to assessment of behaviour in relation to TAB is needed. Patient feedback could be included in TAB which can make it a more reliable tool for assessing a doctor’s behaviour.Further qualitative studies to explore the views and experiences of trainees can help to understand the barriers and attempt to improve the usefulness of the process for the trainees. There are several potential barriers which can subvert the process of MSF by using TAB, and these need to be addressed to make the assessment process more effective and efficient. KEY POINTS
MSF is designed to assess professional behaviour and attitudes
TAB along with Mini-PAT and other tools map MSF principles
The supervisor assumes a key role in giving constructive feedback without demotivating the trainee
Relevant training while giving feedback is critical to the success of MSF
Patient’s involvement in the MSF process could make it more robust.
Leg ulcers are defined as discontinuity of the epidermis and dermis in the lower limb of more than 6 weeks duration 1, 2. They are a common presentation in the elderly population and are associated with a negative impact on the quality of life of patients and they also cause a substantial burden on the health budget 3. Pathogenesis of leg ulceration is heterogeneous 4. Prevention strategies, early identification and proper management are paramount in improving quality of life of patients and reducing costs on an already strained health budget. In this article we review the prevalence of leg ulcers in the elderly people, its common causes and management.
Prevalence The estimated prevalence of leg ulcers in the UK is between 1.5 and 3 per 1,000 population 3.A studies by Moffat et al 2004 showed a prevalence of 0.45 per 1000 which is less than the previously reported figures 5. In the same study leg ulceration was found to be more common in females than males 5. In a systematic review of prevalence studies for leg ulcer, the authors also reported an increase in prevalence of leg ulcers with age and in women 6.The overall prevalence of ulcers is not affected by social class although ulcers tend to take longer to heal in lower socio economic classes 7.Quality of life Several studies have shown that patients with leg ulcers have a poor quality of life compared to age matched controls 8, 9.A systematic review of studies measuring quality of life of patients with leg ulcers showed a negative impact on several domains of quality of life 8. In most of the studies pain was shown to be the major complaint among leg ulcer patients compared to controls, with males experiencing greater pain intensity than female patients. 10, 11 Restricted mobility and sleep disturbance due to pain was also reported in other studies.12 Leg ulcer patients often complain of itchiness, odour and leg swelling 8. In one study, unpleasant odour was reported as causing social embarrassment leading to higher anxiety and depression scores as well as altered body image 12.Reduced mobility due to leg ulceration can restrict working capacity in younger patients13.A negative emotional impact on life with symptoms such as anger, depression, and social isolation was reported by 68% of patients in another study looking at impact of leg ulcers on quality of life 14. Aetiology Several factors contribute to the development of leg ulcers. However majority of ulcers are due to venous insufficiency which accounts for about 80-85% of all cases. 15, 16. Frequency of venous ulcers increases with age as a result of several factors such as immobility and venous disease15. Other risk factors for venous ulceration include obesity, previous deep vein thrombosis, thrombophlebitis, previous fracture, and varicose veins 17.Venous ulcers(also referred to as varicose or stasis ulcer)are commonly found between the malleoli and lower calf and are associated with a shallow base covered with granulation tissue and fibrinoid material, and have irregular margins 16,17. The mechanism of venous ulceration involves initial damage to valves as a result of thrombosis or valve incompetence in varicose veins leading to pooling of blood in lower limbs. Extravasation of red blood cells then follows which causes a local inflammatory reaction and collagen deposition. This impairs the healing process eventually resulting in tissue breakdown and hence ulceration16. Venous ulcers are also associated with symptoms such as oedema, eczema.Arterial ulcers form the second largest group of leg ulcers and account for about 20% of leg ulcers. Atherosclerosis and diabetes are the commonest causes of this group of ulcers. Thrombotic episodes secondary to vasculitis, thromboangitis, and sickle cell disease can also result in arterial ulcers17.Arterial insufficiency causes hypoxia, ischaemia, tissue necrosis and consequently ulceration 18.Arterial ulcers are usually found below the ankle especially on the toes. The ulcers are characteristically small, have steep edges and a dry base. Risk factors for arterial ulcers include conditions that predispose to peripheral vascular disease such as smoking, diabetes, hypertension, hyperlipidaemia and obesity 17. It is worth mentioning that diabetes causes foot ulcers via two mechanisms: ischemia and neuropathy. Neuropathic ulcers are usually found on the plantar surface of metatarsal heads or on the toes. They are a consequence of poor glycaemic control19. The risk of malignancy in chronic leg ulcers is generally believed to be small, but a study by Yang et al 1997 showed a rate of 4.4% in chronic leg ulcers. The diagnosis should be considered in patients with non healing ulcers despite optimum management 20. Chronic inflammatory conditions such as rheumatoid arthritis, inflammatory bowel diseases are also associated with leg ulceration. Rare causes of leg ulceration such as ill fitting shoes have also been reported in literature. In a study looking at complications of ill fitting footwear among 65 elderly patients, foot ulceration was reported by 15% of the patients 21. Although small, this study showed that simple measures such as appropriate foot wear may be useful in preventing foot ulcers. Table 1: summarises causes of ulcers and main characteristics of leg ulcers 16, 17
Aetiology of ulcer
Characteristics
Venous
Between malleoli and lower calf, shallow, irregular margins, granulation base. Oedema, eczema
Arterial
Painful, below ankle distal, especially toes, small, dry baseIntermittent claudication
Vasculitis
Associated with Rheumatoid arthritis, Polyarhritis Nodosa(PAN)
Malignancy
Basal cell , squamaous cell carcinoma, melanoma
Neuropathic
Common in diabetes, wet, deep, sharp borders on pressure points
Management Like all medical conditions, management of leg ulcer should include a detailed history of the onset of the problem (as well as past medical history), examination of the legs and skin, investigations and modalities of treatments. The underlying causes need to be identified as this has crucial implications for management. A medical history suggestive of venous and arterial ulcers have been mentioned above but other factors to consider while assessing leg ulcer are: general health status, cigarette smoking, nutrition, limitation to self care, pedal pulses, Ankle Brachial Pressure Index (ABPI), oedema, limb size and shape, sensation and pain (Table 2). Table 2: Assessment of lower limb ulcers
Patient
History of ulcer developmentPast and current medical problemsGeneral health statusNutritionSocial, occupationMobility problemsLimitation to self careObesityDepression
Skin changes
VenousArterialMalignantAuto immune
Vascular assessment
Pedal pulsesAnkle Brachial Pressure Index
Limb factors
OedemaCircumferencesLymphoedemaOrthopaedic problemsSensation and pain
Examination of the legs and skin identifies markers of underlying pathology. Venous disease may present with some or all of the following: brawny skin, haemosiderin staining, lipodermatosclerosis, atrophie blanche (patchy areas of ischemia),and stasis eczema, while the skin of patients with arterial disease is often shiny, hairless, pale and cool; with thickened nails and changes in foot structure. The absence of venous or arterial signs and symptoms raises the possibility of less common causes of ulceration like: Sun damaged skin, Bowen disease or a history of previous skin cancer treatment is an alert to a malignant lesion. Site & Appearance: Most venous ulcer occur in the “gaiter” area of the leg (i.e. area extending from just above the ankle to below the knee and tends to occur on both lateral and medial aspect of the leg), they are usually superficial with poorly defined margins. The base of the wound is usually red granulation tissue with moderate to high levels of exudates. Arterial ulcers can occur anywhere on the lower leg and may appear in the gaiter region, especially with coexisting venous disease. Many arterial/ischaemic ulcers occur over bony prominences and have a history of pressure related cause. They are often deeper with a punched out appearance and may involve structures such as muscles, tendon and bone in the base. They have sloughy, devitalized tissue in the wound base and low levels of wound exudates. Ulcers occurring in atypical site with an atypical appearance require further investigation to determine the cause. Ulcers with a violaceous (purple) border, inflammation, and extreme pain, may be related to vasculitis problem or underlying connective tissue disorder. Size: Dimensions of the ulcer should be taken at first presentation and fortnightly thereafter and recorded in the notes. This is important as it gives an objective assessment of the effectiveness of the current treatment plan, and modify as necessary22.There are a range of techniques available such as digital photography, ruler based vertical, horizontal and depth measurements and circumferential tracings of wound margins using acetate sheets over cling film .The system chosen needs to reflect consistency, accuracy, and reduced operator error, and also provide visual feedback to the patient. Pain: The level of pain associated with ulcer must be assessed on presentation and at each visit thereafter using a standardized pain scale (0-10). Pain may suggest infection or arterial disease, so careful assessment is required.Surrounding skin area should be observed for the presence of eczema, hyperkeratosis skin, maceration, cellulitis, signs of irritation and scratching which are signs and symptoms associated with underlying venous disease.Assessment should also include palpation of peripheral pulses, regular blood pressure measurement, weight (with reference to a Body Mass Index chart) as well as routine urinalysis (to screen for diabetes). Vascular Assessment: This is mainly carried out by the use of Doppler ultrasound to measure the Ankle Brachial Pressure Index (ABPI).This is mandatory and must be repeated every 3 months. All patients with a non healing wound on the leg of greater than 4-6weeks should have a vascular assessment to eliminate any underlying ischaemic disease22.The result of ABPI are used to determine the likelihood of arterial insufficiency and can be used to guide the management plan, especially in relation to healing potential, referral for vascular assessment and use of appropriate compression bandages. 23 The normal range for ABPI is 0.8-1.2. An ABPI of less than 0.5 or greater than 1.2 needs vascular opinion.Treating the ulcer Many dressing materials are available for the treatment of leg ulcers and there is no adequate evidence from clinical trials to recommend one dressing type over another, but we have to bear in mind few criteria in choosing a particular dressing. The dressing should be low adherent, cost effective and must also be comfortable as well as acceptable for the service user. The choice of product should be determined by the level of exudate. Products which commonly cause skin sensitivity such as those containing lanolin and topical antibiotics should not be used on any service user22. The use of white soft paraffin has been identified as a potential fire hazard risk, hence a water based emollient should be considered as an alternative to a paraffin emollients e.g aqueous cream. Please note water based emollients are not as effective in providing sustained emollient therapy as an ointment and also contain preservatives, which are known potential irritants. Other modalities of treatments of leg ulcers are described below. Compression Therapy The mainstay of treatment of any venous component to ulceration is the application of sustained, graduated compression at therapeutic level 24 .Graduated compression increases venous flow, decreases valvular reflux while walking and increases the effectiveness of the calf muscle pump resulting in a “thinning leg”. The most effective level of compression to overcome venous hypertension has been determined to be around 40mmHg at the ankle 25. Correct application of bandages is essential to avoid pressure ulceration over the bony high points and along the anterior border of the tibia. It is acknowledged that the application of compression bandaging is a specialized skill traditionally undertaken by nurses. The combination of compression bandages used to achieve compression of 40mmHg at the ankle will depend on ankle circumference, according to Laplace’s law, which states that the sub bandage pressure is inversely proportional to the circumference of the limb. A modified compression regimen is necessary when pain is present. This may be achieved by providing periods of relief until pain is controlled or removing the bandage at night when the leg is elevated. Patients with mixed arterial and venous disease may only tolerate up to 20mmHg compression to treat oedema. Bandage choices include short stretch, long stretch, multilayer systems and stockings. A Cochrane Review of compression regimens identified increased healing rates with compression compared to no compression26 . A high compression bandages were better than moderate compression bandages, and that multi-layered bandages were better than single layered bandages. Comparisons between various high compression bandages systems, e.g. 4 layer and short stretch bandages, were unable to find any difference in effectiveness. For venous ulcer with ABPI > 0.8, use 4 layer bandages as per ankle circumference below (Table 3) Ulcers with ABPI between 0.6-0.8: Reduced compression is achieved by omitting the outer cohesive bandage (e.g. Coban) Table 3: Multilayered bandage regime in relation to ankle circumference.
ANKLE CIRCUMFERENCE
BANDAGE REGIME
Up to 18cm
2 or more wool padding1 light stretch bandage(Elastocrepe)1 light elastic bandage 3a(K-plus)1 cohesive bandage 3b(Coban)
Once the ulcer site is well healed, continue with the compression bandages for at least 4 weeks and then maintain compression at a slightly lower level indefinitely as tolerated. It must be replaced annually. Other factors to consider and deal with include: Infection This requires regular cleansing, more frequent changes of dressing (especially if exudates levels are high), topical antimicrobial dressings or systemic antibiotics. Pain Ranging from simple analgesia to potent opioid (depending on severity of pain), non steroidal anti inflammatory drugs may be beneficial. For neuropathic pain, amitriptyline can be started which can be replaced by gabapentin if no improvement. The dose can be titrated upwards. Pressure: Requires pressure relief for the ulcer to heal especially over bony prominences. Larval Therapy Larval therapy has been used for debridement of wounds for many years.27 Debridement is an essential component of wound care as the presence of devitalized tissue can impede the healing process. While the exact mechanism of larval therapy remains unknown, it encompasses three processes: debridement, disinfection and promotion of healing. The beneficial effects of larval therapy were first observed during the Napoleonic war by Larrey, who noted that soldiers whose wounds had become infested with maggots had an improved prognosis 27. During the First World War, Baer documented the successful treatment of leg ulcers and osteomyelitis using larval therapy, and paved the way for further use of it by doctors of that time. However, the development of antibiotics and improvements in surgical techniques reduced larval therapy to a “treatment of last resort”, reserved for the most intractable wounds 28. The emergence of antibiotic- resistant strains of bacteria such as methicillin resistant staphylococcus aureus (MRSA) and the curiosity of researchers has prompted a resurgence of interest in larval therapy. Larval therapy has been employed effectively to treat a wide spectrum of wounds including venous and arterial leg ulcers29.Some of the benefits of larval therapy include: reduction in wound pain and odour, and promoting healing process with relatively few side effects30 .Larval therapy is also reported as being cost-effective in comparison with conventional wound dressings. The use of larval therapy often resulted in quicker healing, and a subsequent reduction of nursing time and materials30. A further advantage of larval therapy is that, as larvae are typically applied for 3 days, wounds are disturbed less frequently than conventional dressings that require changing every 1-2days.In addition to this, a further advantage is that treatment can usually be carried out in outpatient and community settings. A study at an outpatient wound clinic on chronic wounds, of varying aetiologies reported that using larval therapy resulted in a 62% decrease in need of amputation 31. Larvae offer the benefit of eliminating bacteria from the wound through ingestion and subsequent degradation within their intestinal tract. They also act to reduce bacterial activity through the production of inhibitory secretions. The most commonly mentioned disadvantage of larval therapy is the negative perception with which it is regarded by both patients and practitioners because of the unpleasant appearance. The use of “Biobags”, which completely enclose the larvae within a polyvinyl alcohol membrane, has become a popular method of improving the application of this treatment, as larvae are able to feed freely through the open cell polymer. Pain has occasionally been reported by patients, the cause may be the sharp mouth hooks and spicules with which larvae anchor themselves onto tissue. A case history has suggested larval therapy to be contraindicated with fistulae, exposed wounds connecting to vital organs32 because bloodstream infections have been reported with some larvae33.Alteration of the disinfection process appeared to eliminate this problem, and with no further cases of sepsis occurring during the subsequent 12 months The risk of cross-infection by escaped larvae may be greatly reduced through careful dressing. Vacuum Assisted Closure therapy Vacuum Assisted Closure (VAC) therapy involves the application of controlled negative pressure to wounds 34. Negative pressure, as a method of management for difficult to heal wounds, was initially explored in 1970, with the first wound drainage system being introduced in 1989. The use of negative pressure to heal wounds, however, is more commonly associated with the work of Argenta and Morykwas in 1997.35 VAC therapy was designed with the aim of improving healing, decreasing morbidity, and decreasing the cost and length of hospital stay in patients with chronic, non healing wounds. VAC therapy promotes healing in several ways. Firstly, the foam dressing, in combination with adhesive tape, creates an occlusive dressing. This alone prevents dessication and increases the rate at which epithelial tissue is developed, therefore aiding healing times. Occlusive dressing prevents an increase in infection. Secondly, the suction effect and the mechanical forces generated at the interface of the foam work to decrease interstitial fluid accumulation, control wound exudates, stimulate granulation tissue formation, reverse tissue expansion, decrease bacterial colonisation and increase blood flow and dermal perfusion. In summary, VAC therapy aids wound healing by: Maintaining a moist environment Increasing local blood flow Removing wound exudates Promoting granulation tissue formation Reducing infection Exerting mechanical pressuresVAC therapy is suitable for the following wound types/processes 36 Acute (trauma, burns) Chronic (pressure sores, leg ulcers, diabetic ulcers) Surgical (skin grafts, flap surgery, wound bed preparation) Salvage (wound dehiscence, wound infection, post operative sternum infections) Contraindications to the use of vacuum therapy include: wounds with untreated osteomyelitis, grossly infected wounds, when necrotic tissue is present or when there is unspecified disorder of the blood. VAC therapy should also not be used in wounds with malignancy .Dressings should not be placed over any exposed vessels or organs and VAC therapy should be used with caution in patients with active bleeding, difficult wound haemostasis and in patients taking anticoagulants. Skin grafting Skin grafting is the transplanting of skin, and, occasionally, other underlying tissues to another location of the body. It is the only means of reconstructing a defect in the skin, regardless of the cause of the defect 37. Generally, skin grafting is used when, in the opinion of the reconstructive surgeon, other methods of reconstruction such as primary closure, secondary intention healing, or local skin flap are inappropriate, are unavailable or would produce a suboptimal result. Skingraft are divided into 2 major categories: full thickness skin graft (FTSGs) and split thickness skin graft ( STSGs).STSGs may be subdivided into thin (0.008-0.012mm),medium(0.012-0.018mm) and thick (0.018-0.030mm)grafts 37. STSGs are the one used in covering chronic unhealing cutaneous ulcer. Split skin grafting is technically demanding and requires hospital admission25. The discharge from the surface of venous ulcers tends to dislodge continuous sheets of split skin, leaving a choice between mesh and pinch skin grafting. Pinch skin grafting provides epithelial islands, from which epithelial growth may spread outwards as well as inwards from the ulcer margin. Pinch skin grafting has been done by district nurses in the community and has been found to be cost effective accelerating healing when used with multilayer compression bandaging.25 Some contraindications to the use of skin graft generally include: vascular tissues such as exposed bone or cartilage (as this will lead to graft necrosis), uncontrolled bleeding of the recipient because of haematoma and/or seroma formation under the graft compromises graft survival. Venous Surgery Superficial venous surgery has been shown to improve ulcer healing in patients with only superficial venous incompetence38. In patients with no deep reflux on duplex imaging, superficial venous surgery has been shown to reduce long term ulcer recurrence25 . Indications for superficial venous surgery are:
Patient fit for surgery
Sufficient mobility to activate calf muscle pump
Prepared to attend hospital for investigation and surgery
Obesity controlled (BMI <30)
Superficial venous incompetence
There are few other modalities of treatment of leg ulcers currently in use, although no strong evidence has been found to show they really improve healing. Intermittent Pneumatic Compression Intermittent pneumatic compression (IPC) is a mechanical method of delivering compression to swollen limbs that can be used to treat venous leg ulcers and limb swelling due to lymphoedema.It uses an air pump to inflate and deflate an airtight bag wrapped around the leg.39 However, review of trials found conflicting evidence about whether it is better than compression bandages. It may increase healing compared with no compression but it is unclear whether it improves healing when added to treatment with bandages. Electromagnetic Therapy Electromagnetic therapy involves the use of electromagnetic, microwave, or infrared energy to diagnose or treat an illness by detecting and correcting imbalances in the body's energy fields. Electronic devices that emit some form of low-voltage electrical current or radio frequency are often involved. It has been used in the treatment of chronic diseases like venous leg ulcers. Cochrane wound group conducted trials on several occasions comparing electromagnetic therapy with other treatments and up till now, there is no reliable evidence of benefit of electromagnetic therapy in the healing of venous leg ulcers.40 Further research is still needed.Oral Zinc Supplement Leg ulcers may take long time to heal despite good wound care. This may be due to poor nutrition which reduces the ability of the body to repair itself. Minerals such as zinc are necessary for good healing and so it has been thought that taking zinc sulphate tablets might aid healing of ulcers especially if patients were found to have low baseline Zinc level. Few trials were found where zinc was used to treat leg ulcers but all were too small to pick up on any benefit, if such a benefit exists. In addition, the quality of those trials was mediocre. On the basis of the evidence available so far, it appears that taking zinc tablets does not improve leg ulcer healing, however more good quality trials are needed.41Laser Therapy Low level laser therapy (LLLT) refers to the use of red beam or near infrared laser with a wavelength between 600and 1000nm power from 5-500 milliwatts.It is also referred to as cold laser therapy, low power laser therapy (LPLT), low intensity laser and low energy laser therapy. The exact effect of its mechanism is unknown; however, hypotheses have included improved cellular repair and stimulation of the immune, lymphatic and vascular system. Several randomised controlled trials involving patients with venous ulcers failed to demonstrate any significant benefits of LLLT when compared to standard treatment methods or placebos.42Hyperbaric Oxygen Therapy Oxygen is one of the most versatile and powerful agents available to the modern medical practitioner. The therapeutic use of oxygen under pressure is known as hyperbaric oxygen therapy (HBO2) and has been used to assist wound healing for almost 40 years. HBO2 has several specific biological actions which can enhance wound healing processes .These include: Hyper-oxygenation of tissue, vasoconstriction, down regulation of inflammatory cytokines, up-regulation of growth factors, antibacterial effects, potentiation of antibiotics, and leukocyte effects.43 Systemic oxygen can be administered via 2 basic chambers: Type A( multiplace) and Type B(monoplace). Both types can be used for routine wound care, treatment of most dive injuries, and treatment of patients who are ventilated or in critical care.HBO2 is a relatively safe non-invasive therapy. Side effects include middle ear and pulmonary barotraumas and myopia. Contraindications include poor cardiac output and severe obstructive pulmonary disease. Conclusion In this article, we have been able to show that leg ulcers are a common presentation in the elderly population and have negative impact on the quality of life of affected patients. It has been found to be more common in females. Most leg ulcers (about 80-85%) are caused by venous insufficiency, followed by arterial ulcers. A comprehensive assessment of the patient, skin, vascular status, limb and ulcer is required to determine aetiology and to formulate an appropriate management plan as described above. Several researches are still going on other modalities of treatment of leg ulcers. However, all patients should be provided with both verbal and written information to help them understand their condition and treatments they receive, as this will enable them to better understand their conditions, and will support concordance between patients and staff.
Spasticity is a common symptom seen as a consequence of an injury to the brain (stroke, trauma, hypoxia, infection, cerebral palsy and post surgery), spinal cord injury or multiple sclerosis. It is a complex problem, which can cause profound disability alone or in combination with other features of upper motor neuron syndromes (figure 1). The word “spasticity” is derived from the Greek word “spasticus”, which means “To pull or To Tug”. Spasticity is defined1 as “Disordered sensori-motor control, resulting from an upper motor neuron lesion, presenting as intermittent or sustained involuntary activation of muscles”. Simply stated, spasticity is stiffness of muscles that occurs after injury to the spinal cord or brain. Awareness of the implications and associated symptoms can minimise development of long term secondary complications (table 1). The impact of spasticity can be devastating. If not managed early and appropriately it may result in progressive disability, resulting in secondary complications such as contractures and pressure sores. This significant impact has ensured that spasticity management is a prominent feature in the national management guidelines for long term neurological conditions, promoting coordination of care between primary, secondary and social care providers. Symptoms Spasticity can range from mild muscle stiffness to severe, painful and uncontrollable muscle spasms. It is associated with both positive and negative components of upper motor neuron syndromes. Positive components include muscle overactivity, flexor and extensor spasm, hyperreflexia, athetosis, spastic dystonia, clonus, and an extensor plantar response. Common negative symptoms comprise weakness/ paralysis, early hypotonia, fatigue and loss of dexterity. Spasticity can be distinguished from rigidity by its dependence on the speed of muscle stretch and characteristic distribution in antigravity muscle groups. Spasticity does not always cause harm and can occasionally assist in the rehabilitation process by enabling a patient to stand when their limb weakness would not otherwise allow it Table 1 Clinical and functional problems associated with severe Spasticity
Physical
Emotional / social
·Non- specific pain·Discomfort·Painful muscle spasm·Difficulties with activities of daily living. e.g. washing, dressing, eating, toileting, maintaining hygiene, sexual activity·Problems with posture and mobility·Physical deformity and longterm contracture·Pressure ulcers
·Emotional e.g. low mood, distorted self image, impaired motivation·Impact on fulfilment of life roles as a partner or a parent·Sleep disturbance – due to pain and discomfort·Vocational- impact on employment or education·Social isolation – due to restricted mobility
Figure 1 Vicious cycle of spasticity Assessment of spasticityBefore any intervention is undertaken to modulate hypertonicity, it is important to attempt to assess the severity of spasticity. Many grading scales are used to quantify spasticity. These address the degree of muscle tone, the frequency of spontaneous spasms and the extent of hyperreflexia. Goniometry, Ashworth scale, Tardieu Scales, Goal attainment scale are only a few of these scales. One of the most widely used scales is the modified Ashworth scale2. Table 2 Modified Ashworth scale
4 Rigid extremity 3 Loss of full joint movement, difficult movement, considerable tone2 Full joint/ limb movement, but more increase in tone, limb still easily moved.1+ Slight increase in tone, catch and resistance through out range of movement1 Slight increase in tone, catch or minimal resistance at end of range of movement0 no increase in tone
It is also important to remember that not every “tight” muscle is spastic. The clinically detectable increase in muscle tone may be due to spasticity, rigidity or a fixed muscle contracture.Management The key to successful spasticity management is education of the patient and carers with both verbal and written information. This allows them to understand, appreciate and be fully involved in the management plan. All patients with spasticity should be followed up by a coordinated multidisciplinary team, which allows more timely intervention and close monitor of the progress. Liaison between health and social services in both primary and secondary care is essential in long term management. This helps to deliver a more consistent approach to the individual over time (figure 2). Table 3 Aims of spasticity management.
1.Improve function- mobility , dexterity2.Symptom relief-· Ease pain- muscle shortening, tendon pain, postural effects· Decrease spasms· Orthotic wearing3. Postural- Body image4. Decrease carer burden- Care and hygiene, positioning, dressing5. Optimise service responses- to avoid unnecessary treatments, facilitate other therapy, delay/prevent surgery
The first step in the management of spasticity is to identify the key aims and realistic goals of therapy. Understanding the underlying pathology and possible prognosis is helpful in planning these goals (table 3). Other key points to consider are: · Identification and management of any trigger or aggravating factors-Initial assessment should exclude any co morbidity that may worsen spasticity such as pressure sores, chronic pain, infection (commonly urinary tract infection), constipation or in-growing toe nails.· Instigation of an effective and realistic physical programme including attention to posture and positioning Figure 2 Approach to spasticity assessmentComprehensive assessment of spasticityRecognition of underlying provocative factorsImpact on the individualMeasurement of spasticityPotential goals- individualised and person focussed Initial approachEducation to patient/ and their family /carersMultidisciplinary team assessmentIdentification of clear treatment goalsEstablish mechanism for monitoring and review Management Manage triggering factorsBalance between positioning and movementPosture and seatingPhysical therapy and active exercise programmeSplinting and use of orthoticsPharmacotherapy A) Physical modalities· Stretching- this intervention has the benefit of being benign and non-invasive. Maintaining muscle length through passive or active exerciseand stretching regimens including standing or splinting canbe key to managing spasticity both in the short and the longterm.· Cooling of muscles- this inhibits mono synaptic stretch reflex and lowers the receptor’s sensitivity, different techniques such as quick icing and evaporating spray like ethyl chloride are occasionally used.· Heat-heat may increase the elasticity of the muscles. Techniques used include ultrasound, fluidotherapy, paraffin, superficial heat and whirlpools. These techniques should be combined with stretching and exercise.· Orthosis/equipment/ aids – an orthosis or splint is an external device designed to apply, distribute or remove forces to or from the body in a controlled manner to control body motions and /or alter the shape of body tissues. E.g. ankle foot orthosis, insoles, ankle supports, wrist/ hand/ elbow splints, knee splints, spinal brace, hip brace, neck collar. Some equipment can also aid positioning e.g. T roles, wedges, cushions and foot straps.These are usually used in combination with other modalities like botox therapy. Attention to posture and positioning, whichmay include the provision and regular review of seating systems,is paramount in managing severe spasticity· Massage– althoughvarious techniques are in use there is no evidence to support this· Dynamic physiotherapy technique- many schools of physiotherapy claim that particular technique has antispastic and functional benefits, particularly for the more mobile person. E.g. Bobath technique, proprioceptive neuromuscular facilitation, Brunnstrom technique. B) Electrical therapy· Functional Electrical stimulation –Thisis an adjunct to physiotherapythat can be of benefit to selected individuals who are predominantlyaffected by upper motor neuron pathologies resulting in a foot drop. Randomised controlled trial by Burridge et al in patients following stroke found that the use of functional electrical stimulation in combination with physiotherapy was statistically superior to physiotherapy alone3· Transcutaneous electrical nerve stimulation – this has been found to reduce spasticitythrough its nociceptive action and reduction of pain.C) PharmacologicalMedicationshould always be used as adjunct to good general management and education. Identification of treatment goals will help optimise drug therapy not only in terms of choice of agent, but also in timing and dose. Aims of medication should be to improve function or relieve troublesome symptoms rather than to simply reduce the degree of spasticity. Table 4 Useful things to remember to optimise medication effects
1. Clear written/verbal information for patients about effects/adverse symptoms of drugs 2. Clear treatment goals3. Detailed drug history- Review of other medication and potential drug interaction4. Appropriate form of drug e.g. liquid preparation if swallowing difficulties5. Regular review of efficacy and side effects6. Aids to help administer drugs e.g. dossette box, timer to remind7. “Start low and go slow” to avoid deleterious effects on function or unwanted side effects8. Combination of drugs to obtain synergistic action
Table 5 Different methods of delivery of medication
·Enteral – orally or via PEG e.g. baclofen, benzodiazepins, dantrolene, clonidine, tizanidine, gabapentin·Transdermal system e.g. catapress TTS·Intrathecal e.g. baclofen pump (other drugs used alone or in combination intrathecally include clonidine, morphine, fentanyl, midazolam, lidocaine)·Intra muscular/ focal injection e.g. botulinum toxin·Nerve blocks e.g. Phenol, Ethanol
The oral agentsAlthough different categories of drugs are available, those most commonly used to treat spasticity are baclofen,tizanidine, benzodiazepines, dantrolene, and gabapentin4, 5, 6. Different agents act through different mechanisms (table 6 and 7) for e.g. GABA-like (baclofen, benzodiazepine), central alpha 2 agonists (tizanidine, clonidine) and peripheral anti-spastics (dantrolene). Antispastic drugs act in the CNS either by suppression of excitation (glutamate), enhancement of inhibition (GABA, glycine) or a combination of the two. Table 6 Mechanism of action of commonly used oral antispasticity medication
Drugs acting on
Drugs
GABA- ergic system
baclofen, benzodiazepines, piracetam, progabide
Ion flux
dantrolene sodium, lamotrigine, riluzole
Monoamines
tizanidine, clonidine, thymoxamine, beta blockers, and cyproheptadine
Excitatory amino acids
orphenadrine citrate, cannabinoids, inhibitory neuromediators and other miscellaneous agents.
Baclofen remains the most commonly used anti-spastic agent. The preferential indication is spasticity caused by spinal cord disease especially in multiple sclerosis. Many studies including the pilot study by Scheinberg et al 7 demonstrated that oral baclofen has an effect beyond placebo in improving goal-oriented tasks (such as transfers), in children with spastic quadriplegic cerebral palsy. In open-label studies of oral baclofen, the drug improved spasticity in 70-87 per cent of patients; additionally improvement in spasms was reported in 75-96 per cent of patients. In double-blind, crossover, placebo-controlled trials, baclofen was reported to be effective, producing statistically significant improvements in spasticity8.The main adverse effects of oral baclofen include sedation or somnolence, excessive weakness, vertigo and psychological disturbances. The incidence of adverse effects is reported to range from 10 to 75 per cent. The majority of adverse effects are not severe; most are dose related, transient and/or reversible. The main risks of oral baclofen administration are related to withdrawal; seizures, psychic symptoms and hyperthermia. These symptoms improve after the reintroduction of baclofen, usually without sequelae. When not related to withdrawal, these symptoms mainly present in patients with brain damage and in the elderly. The limited data on baclofen toxicity in patients with renal disease suggest that administration of the drug in these persons may carry an unnecessarily high risk. Tizanidine is an efficient and well tolerated antispastic. It is predominantlyan alpha 2 agonist and thus decreases presynaptic activity of theexcitatory interneurones. There is a large body of evidence for the effective use of tizanidine monotherapy in the management of spasticity 15. Tizanidine is the antispasticity drug that has been most widely compared with oral baclofen. Studies have generally found the two drugs to have equivalent efficacy, although tizanidine has better tolerability; in particular weakness was reported to occur less frequently with tizanidine than with baclofen.Dantrolene has a peripheral mechanism of action and acts primarily on muscle through inhibitingcalcium release from the sarcoplasmic reticulum. It decreasesthe excitation–coupling reaction involved in muscle contraction and can be prescribed in the different forms of spasticity. The efficacy of benzodiazepines (diazepam, tetrazepam, clonazepam) is comparable with baclofen. Although there is no evidence to suggest any difference in effectiveness between them, diazepam and dantrolene are associated with more side effects than baclofen and tizanidine. There are other compounds with anti-spastic properties (gabapentine, cyproheptadine, piracetam). Their advantage is
Table 7
Drug
Dosage
Doses per day
Mechanism of action
Common side effects
Initial dosage
Maximum dosage
Baclofen
5mg x3
90mg
4
GABA ergic
Seizure, Sedation, Dizziness, GI disturbances, psychosis, Muscle weakness
rather limited when used alone. Generally, they are administrated in combination with usual anti-spastic drugs. A few short term trials have trialed gabapentin with good results 19 .Pregabalin may be of value as a systemic agent in the treatment of spasticity, although properly controlled studies with clearly defined outcome measures are required to confirm this finding 22. The Sativex Spasticity in MS Study Group23 concluded that oromucosal whole plant cannabis-based medicine (CBM) containing delta-9 tetrahydrocannabinol (THC) and cannabidiol (CBD) may represent a useful new agent for treatment of the symptomatic relief of spasticity in MS. Intrathecal pump· Baclofen- If oral drug treatment is inadequate in controlling lower limb spasticity or is not tolerated, intrathecal delivery of baclofen should be considered. This has been found to be a cost-effective strategy when compared to conventional medical management alone by Bensmail et al 20 .The benefits of continuous intrathecal baclofen infusion have been demonstrated in more than 80 percent and over 65 percent of patients report an improvement in tone and spasms respectively. The main risks of intrathecal baclofen infusion are symptoms related to overdose or withdrawal. These are mostly related to catheter disruption, failure to refill the pump reservoir or failure of the pump's power source. Abrupt disruption of intrathecal baclofen can be a serious scenario with continuous spasms, tremors, temperature elevation, seizure and death having been reported.· Phenol- As phenol is a destructive agent which indiscriminately damagesmotor and sensory nerves, it is reserved for those individualswho do not have any functional movement in the legs, who havelost bladder and bowel function and who have impaired leg sensation. Intrathecal phenol can be an effective treatmentwhich, though it requires expert administration, does not havethe long term maintenance or cost issues that are associated with intrathecalbaclofen treatment. The effect of a single injection often lastsmany months and can be repeated if necessary 24. D) Nerve blockPeripheral nerve blockade/ Regional blocks/ Neurolytic blockade25 are another therapeutic possibility in the treatment of spasticity. This can be done with the help of fluoroscopy or nerve stimulation. Chemical neurolysis by phenol/ alcohol is irreversible and can be used at several sites. Blocks are applied most often to 4 peripheral sites: the pectoral nerve loop, median, obturator, and tibial nerves. The main indication is debilitating or painful spasticity. Peripheral blocks with local anaesthetics are used as tests to mimic the effects of motor blocks and determine their potential adverse effects. Peripheral neurolytic blocks are easy to perform, effective, and inexpensive30.E) Botulinum toxin injectionBotulinum toxin is the most widely used treatment for focal spasticity27,28,29. The effect of the toxin is to inhibit the release of acetylcholine at the neuromuscular junction. The clinical effect of injecting botulinum toxin is reversible due to nerve sprouting and muscle reinnervation, leading to functional recovery of the muscle in a few months. It is essential that botulinum toxin injections are given in conjunction with physiotherapy in order to obtain the maximum benefit. The toxin is injected directly into the targeted muscle and an effect can be noticed from as early as 2-3 days with a maximum effect seen by about 3 weeks, lasting at least 3 months. As it is not a permanent treatment it may have to be repeated after a few months.Many randomisedcontrolled trials show that botulinum toxin is effectivein reducing muscle tone in various conditions 28, 29. Brashear and colleaguesdemonstrated a reduction in spasticity in the wrist andfingers of patients following stroke with the use of botulinum toxin, together with an improvementin their disability assessment scale29.E) Surgical techniqueMost surgical procedures are irreversible. This means that realistic goal setting between the health care provider, family and patient is critical. Neurosurgical techniques have been proven useful in conditions like cerebral palsy 32, 33.· Neurosurgical techniques- Anterior and posterior rhizotomy, peripheral neurotomy 31, Drezotomy, percutaneous radiofrequency rhizotomy, spinal cord and deep cerebellar stimulation of the superior cerebellar peduncle 32, functional neurosurgery 33· Orthopaedic procedures- directly act on muscles and tendons e.g. lengthening operation, tenotomy, neurectomies, and transfer of tendons.
Key Points to remember1.Spasticity management is more effective in multidisciplinary settings2.Early multidisciplinary approach and goal setting is crucial3.Education and clear communication between patients, carers and health care providers is essential4.Early intervention and optimal therapy prevents long term complications.5.Focal spasticity responds well to botulinum toxin injection, while generalised spasticity needs oral/ intrathecal medications
Introduction.
This legislation is based on rules established by case law about how to work with people who lack capacity (either on a temporary or permanent basis).
The Act provides a definition of capacity, a functional test for capacity (see Box 1) and a checklist for Best Interest decision making which are under pinned by five key principles (See box 2). The Act is supported by a Code of Practice.
The Act, which applies to all adults aged 16 years or over (with some exceptions), provides a clear definition of incapacity, and for deciding if a person lacks capacity in respect of a particular matter.
“A person lacks capacity in relation to a decision or proposed intervention if, at the material time, he is unable to make a decision for himself in relation to the matter or proposed intervention because of an impairment of, or a disturbance in the functioning of the mind or brain. It does not matter whether the impairment or disturbance is permanent or temporary.” (S2 (1) and (2) MCA 2005.
Box 1. Testing Capacity
The responsibility for testing capacity rests with the person who wishes to make a decision on behalf of someone who lacks capacity.
The functional test of capacity:
Does the person have an impairment or disturbance in the functioning of his mind or brain?
Does the impairment or disturbance make the person unable to
A person is unable to take a decision for himself/herself if he/she is unable to:
Understand the information relevant to that decision;
Retain that information long enough to reach a decision;
Use or weigh that information as part of the process of making the decision; or
Communicate his decision (whether by talking, using sign language, visual aids or any other means).
Box 2. Principles (based on section 1 MCA 2005)
Best interests always.
Less restrictive care provision option.
Encourage individual to make own decisions.
Eccentric decisions are OK.
Presume capacity always.
It is important to note that the decision is always ‘time specific’ and ‘issue specific’. It is also a test applied both to people with temporary or fluctuating capacity (such as people experiencing mental ill-health) and those whose decision making ability is permanently impaired (such as people with a learning disability)
The Act starts from the presumption that those we work with do have capacity, and requires staff to involve them as much as possible in their own treatment and care including when there is evidence that they lack capacity in a particular matter.
The Act also introduces a statutory right to advocacy for those lacking capacity and “unbefriended” through the Independent Mental Capacity Advocacy Service (IMCA), Lasting Powers of Attorney for health and welfare and property and finance and two new criminal offences, i.e. “the wilful neglect or ill treatment of a person lacking capacity” (S 44 MCA 2005.)
The MCA will also apply when someone is detained under the Mental Health Act 1983. For example if the person lacks capacity to consent to treatment for a physical health issue rather than treatment related to mental disorder.
The Act has introduced safeguards for medical practitioners when working with advanced decisions made by people in advance for how they wish to be treated when or if they lose capacity in the future.
Best Interests Check List.
The best interests checklist represents the issues that decision makers must consider when decisions or interventions are made on behalf of someone who lacks capacity, if the decisions (and the decision maker) are to be protected by the MCA
The checklist items include that the decision maker:-
· Must not make their judgement based merely on the person’s age, appearance, condition (or diagnosis);
· Must take into account whether the person is likely to regain capacity with regard to the decision in hand, and whether the decision can wait;
· Must as far as reasonably practicable, ‘permit and encourage’ the person to communicate, including by acting to improve his or her ability to communicate (for example, by using an advocate);
· Must not, where the decision relates to life sustaining treatment, be motivated by a desire to bring about the relevant person’s death;
· Must so far as is possible consider the person’s past wishes and any preferences (particularly when written down) stated by him or her when they had capacity;
· Must take account of the beliefs and values that would have been likely to influence the person’s decision had they had capacity;
· Must, if practical and appropriate, consult anyone previously named by the patient as someone who should be consulted, any carers, anyone who has a relevant lasting power of attorney – a ‘donee’ (remembering that there are two kinds of LPA – (i) personal welfare, and (ii) property and affairs), and any appointed court deputy about their views concerning what would be in the person’s best interests. Protection from liability offered by Section 5 of the Mental Capacity Act
The MCA provides legal protection for people who need to intervene in the lives of people who lack capacity so that they are able to make a decision on that person’s behalf, or provide the care the person needs, as long as they have a reasonable belief that the person lacks capacity to make the particular decision and they are working in the person’s best interests.
Generally, however, protection is available as long as:-
· Reasonable steps have been taken to gain permission from the person concerned;
· The decision maker is reasonably sure the person lacks the capacity to make a particular decision;
· The decision maker is working in their best interests, and before making the intervention you have considered whether there is a “less restrictive’ option than the one proposed, and only ruled it out because it is less effective than the one you are now taking;
· Restraint if needed, is a proportionate response to the risk of harm if no action is taken;
· The action doesn’t amount to a deprivation of liberty, or conflict with an advance decision made by the person, their LPA or a Deputy;
· The decision maker is spending money to buy goods or pay for services that are in the person’s best interests and appropriate authority has been sought.
In a medical context, this could be helpful on a day-to-day basis, or to deal with an emergency situation where the Mental Health Act does not apply as illustrated in the example, taken form the Code of Practice, in box 3 below.
Box 3.
Example:You are called for advice by a local GP. She is with a patient in her home and the ambulance service is in attendance. The patient is dehydrated, and has a suspected UTI (urinary tract infection). The patient has become angry and belligerent at the idea that she needs admission to hospital and is refusing to go. She says that the doctor is in league with her neighbours and they intend to defraud her of her savings the moment she is out of the house. The ambulance staff refuse to intervene because they say it would contravene the woman’s human rights. The GP is considering asking for a Mental Health Act assessment. She says that, because of the advanced age and presentation of the patient, it is too risky to leave her at home. She confirms that she feels the woman lacks the capacity to take the decision about whether or not hospital admission is necessary because of the acute confusional state brought on by the dehydration and UTI.
You are able to advise the GP and the ambulance staff that, in this situation, the Mental Health Act may not be needed as their intervention would be covered by the Mental Capacity Act. The ambulance staff will be covered by sections 5 and 6 of the MCA, as long as their use of force in taking the woman to A&E is proportionate to the risks that staying at home poses to her. They need to be convinced themselves (as the professionals undertaking the act) that the actions would be in her ‘best interests’. The ambulance staff return to talk to the woman, and coax her to sit on their ‘lifting chair’. They are then able to wrap her securely in a blanket, with straps around her, so that they are able to carry her safely to the waiting ambulance despite her vocal protests.
Limitations to Section 5 by Section 6 Mental Capacity Act 2005.
It is important to recognise that section 5 of the Act does not offer practitioners total freedom from liability in providing care or treatment.
· Life-changing events: decisions about life-changing events, such as changes in residence and serious medical treatment will only be covered under Section 5 if the decision makers firstly consult all appropriate parties, and secondly consider whether there is a less restrictive way in which the care needed can be given. If there are no families or friends that professionals can consult in these specific circumstances, or if the decision maker deems the family member or friends “inappropriate", an Independent Mental Capacity Advocate (IMCA) must be instructed to support and represent the person whilst their best interests are being determined.
· The use of force, and depriving people of their Liberty: doctors and other professionals will continue to be protected by the law where, in an urgent situation, it is necessary to restrain or restrict a person who lacks capacity in order to protect them from harm. The force used must be proportionate to the risks involved. However, this protection has a ‘time limit’. Where restraint is needed on an ongoing basis (and restraint can mean the use of medication, or making a decision or making it known to a patient that they would be prevented from leaving) professionals involved won’t necessarily be protected by the MCA – this is where the Deprivation of Liberty Safeguards become important. Safeguards for patients: making decisions in advance Advance decisions to refuse medical treatment
People can now make advanced decisions to refuse treatment, provided that the decisions were made when the person had the capacity to make them.
To make a valid advance decision, a person must:
· Be 18 years or older
· Have capacity to make the specific decision
· Make a decision that is applicable (i.e. specific to the care and treatment they want to refuse and the circumstances in which it will be refused )
The decision doesn’t need to be in writing, unless it relates to life sustaining treatment – in which case it must be in writing, and witnessed.
An advanced decision becomes valid and applicable when all of the conditions described within it are present.
If Doctors are not informed about the existence of an advanced decision then they are expected to treat someone with that person’s bet interests in mind.
Lasting Powers of Attorney and Deputies from the Court of Protection
The MCA allows people to make arrangements for others to make decisions on their behalf when or if they lack capacity.
Lasting Powers of Attorney (LPAs) – The Act allows a person to appoint an attorney to act on their behalf if they should lose capacity in the future. The Act also allows people to empower an attorney to make health and welfare decisions, as well as financial & property decisions (a LPA for finance and property can be used whilst a person still has capacity, if the donee gives specific instruction). Before it can be used a LPA must be registered with the Office of the Public Guardian (see below).
Court appointed deputies – The Act provides for a system of court appointed deputies to replace the previous system of receivership in the “old” Court of Protection. Deputies will be able to be appointed to take decisions on welfare, healthcare and financial matters as authorised by the new Court of Protection (see below) but will not be able to refuse consent to life-sustaining treatment. They will only be appointed if the Court cannot make a one-off decision to resolve the issues.
A Court of Protection – The new Court has jurisdiction relating to the whole Act. It has its own procedures and nominated judges. It is able to make declarations, decisions and orders affecting people who lack capacity and make decisions for (or appoint deputies to make decisions on behalf of) people lacking capacity. It deals with decisions concerning both property and affairs, as well as health and welfare decisions.
A new Public Guardian – The Public Guardian has several duties under the Act and will be supported in carrying these out by an Office of the Public Guardian (OPG). The Public Guardian and his staff will be the registering authority for LPAs and deputies. They will supervise deputies appointed by the Court and provide information to help the Court make decisions. The OPG runs threes registers for Lasting Powers of Attorney, Enduring Powers of Attorney and Deputies; this information is available to members of the public. The OPG will also work together with other agencies, such as the police and social services, to respond to any concerns raised about the way in which an attorney or deputy is operating.
Independent Mental Capacity Advocate (IMCA) – An IMCA is someone instructed to support a person who lacks capacity but has no one to speak for him or her, such as family or friends , or if family or friends are present but considered “inappropriate” to assist in the process. IMCAs must be involved where decisions are being made about serious medical treatment or a change in the person’s accommodation where it is provided, or arranged, by the National Health Service or a local authority, and may be involved in abuse cases. The IMCA makes representations about the person’s wishes, feelings, beliefs and values, at the same time as bringing to the attention of the decision-maker all factors that are relevant to the decision. The IMCA can challenge the decision-maker on behalf of the person lacking capacity if necessary; challenges can be made via the Court of Protection or Judicial Review process. However, it is still up to the decision maker to consider what they believe is in the person’s best interests.
Key Concepts for doctors:
Lack of capacity in one area can’t be assumed to mean lack of capacity in another – and patients should be as involved as possible in all decisions made about their treatment.
Where it is proposed that a person move permanently into residential or nursing care, or serious medical treatment is proposed for someone who lacks capacity, the person’s relatives must be consulted about what they believe the person’s views about this would be, and whether the move or treatment would be in their best interest. If there are no relatives, an IMCA must be consulted.
The MCA s5 protects staff from liability as long as they have a reasonable belief that a person lacks capacity, and any force used in an urgent situation is proportionate to the risks that would fall to that person if they were not restrained. Where care needs to be provided in such a restricted way that it amounts to a ‘derivation of liberty’, this needs to be authorised. From April 09, the Deprivation of Liberty Safeguards may provide the authority needed to detain someone that is unable to consent to care or treatment being provided in a registered care home or hospital setting. (The Deprivation of Liberty Safeguards are the Government’s response to the European Court of Human Rights’ requirement that the so called “Bournewood Gap” be dealt with in British Law.)
Where staff become aware that a patient has made an advance decision refusing a particular treatment, that refusal has the same force as if the patient where making it contemporaneously, i.e. the medical treatment could not be given unless the doctor concerned was happy either that the patient did not have capacity when the decision was made, or that they did not intend the decision to have effect in the current circumstances.
Conclusion.
Anecdotally, medical practitioners appear to have been slow to make use of the powers and safeguards provided by the MCA. Relatively small numbers of referrals have been made to the IMCA services nationally to support those people that lack capacity and are “unbefriended” in the decision making processes around serious medical treatment. Only 671 eligible referrals were received by IMCA services in England and Wales in 2007/2008. (First Annual Report of the IMCA Service, July 2008). Could it be that an assumption is being made that the IMCA service may be seen more of a hindrance than a help, rather than a safeguard for the patient, in providing care and treatment?
The Act requires professionals to “presume capacity” rather than incapacity, for most professionals this is a challenge that we often fail to meet. It is easier to work with a presumption of incapacity and to act in that person’s best interest rather than take the time to “evidence” their capacity in relation to a variety of decisions that may need to be made.
The Act’s two new criminal offences have resulted in a small number of prosecutions to date. These prosecutions have tended to be brought against staff providing care in care homes or domiciliary settings rather than in hospital or other medial settings. Does this mean that staff working in hospitals or medical settings provide better care?
The Civil Rights Act 1964 remains of the greatest achievements in United States (US) history. It had implications internationally, making racial discrimination illegal, but its effectiveness in the employment domain remains contestable 2. The worldwide existence of workplace racism has attracted controversy and this is drawn out by psychiatry’s attempt to understand the nature of the problem 3. Discrimination at work, based on a person’s race, comes in different guises and can have negative consequences on both individuals and organisations 1. Despite legislation to protect individuals substantial progress needs to be made to eradicate the problem.
What is racism?
The concepts of “race”, “ethnicity” and “racism” are explained in figure 1.
Figure 1: Definition of race, ethnicity and racism 4, 5
Race
The group a person belongs to as a result of a mix of physical features, ancestry, and geographical origins, as identified by others or, increasingly, as self-identified. The importance of social factors in the creation and perpetuation of racial categories has led to a broadening of the concept to include social and political heritage, making its usage similar to ethnicity. Race and ethnicity are increasingly used synonymously.
Ethnicity
The group you belong to as a result of a mix of cultural factors that include language, diet, religion, ancestry, and race.
Racism
A belief that some races or ethnic groups are superior to others, used to devise and justify actions that create inequality between racial groups.
Racism is a social process associated with “overt and covert forceful establishment and maintenance of power by one social group over another” 3.
Racism can be seen as a misuse of power and, even today, power relations are signified by subtle cultural rules that perpetuate racial inequality 6.
What are the origins of racism?
That some races are superior to others has origins from the 19th century 5. The history of racism has stimulated considerable debate in understanding racism.
Racism may have origins in experiences derived from, what is known in analytical psychology as, the collective and personal unconscious. The personal unconscious arises from the lifetime experiences of the individual. This is distinct from the “collective unconscious” which psychiatrist Carl Jung described to represent a form of the unconscious common to mankind as a whole and originating in the inherited structure of the brain 7. This contains inherited primitive cultural and racial elements. Both the personal and collective unconscious, made from our individual and ancestral experiences respectively, may account for the manifestation of racism in society today.
In recent times the experience of overt racial bigotry and prejudice is seldom seen 8. Nevertheless discrimination against members of a social group may persist because it is so deeply entrenched within society, by the personal and collective unconscious, that it becomes the automatic response even when no conscious intent is present 9. “Everyday discrimination” is the discreet, pervasive discriminatory acts experienced by stigmatised groups on a daily basis 10, and highlights the modern perspective that racism is subtle.
The subtlety of racism
“Like a virus that has mutated, racism has evolved into a new form that is difficult to recognise and harder to combat” 8
As blatant forms of racism become extinguished, particularly in the current climate of political correctness, unconscious racial biases in subtle forms, known as ambivalent or modern racism 10, are appearing. This has been referred to as aversive racism occurring in people who possess strong egalitarian values, and who believe they are not prejudiced, but have negative racial feelings and beliefs that they are unaware of 8. These feelings and beliefs are rooted in the normal psychological processes of social categorisation, satisfaction of basic needs for power and control, and socio-cultural influences 8. The ambivalence involving positive and negative feelings creates a psychological tension that leads to an inconsistent pattern in their behaviour 8.
The cumulative effects of unpredictable and seemingly trivial behaviour such as avoidance of ethnic minorities, closed and unfriendly verbal and non verbal communication, and failure to provide assistance, is more damaging 10. Apparently harmless interactions, including racist assumptions and questioning about where somebody is from, also convey messages about marginality and not belonging 11. This subtle racism may contribute to the racism perceived by minority groups in higher status professions and organisations.
Does racism exist in healthcare organisations?
“American Medical Association apologizes for racism in medicine” (10th July 2008) 12.
This admission by the American Medical Association, of racial discriminatory practices against African-American physicians, reflects the recognition of racism in other western countries.
In the United Kingdom (UK) racism has been revealed in public institutions such as the metropolitan police 13 and widely reported in the nursing profession 14,15 within the National Health Service (NHS). Trevor Phillips, chairman of the Equality and Human Rights Commission, referred to the “snowy peaks of the NHS” 16 with a large number of ethnic minorities at the base. Less than 10% of senior managers and 1% of chief executives are from ethnic minority background 17. There is a “glass ceiling” 18 preventing promotion and black and minority ethnic (BME) managers feel they have to work twice as hard and have twice as many qualifications to succeed 19.
Since 2000, after a survey commissioned by Department of Health (DOH) reported that half of front-line NHS BME staff had been victims of racial harassment in the previous 12 months 20, reports of racism in healthcare have increased. In 2001 a Kings Fund report, “Racism in Medicine” 21, generated powerful debate after finding that bullying and discrimination were a daily fact of life for black and Asian doctors. Then in 2003 a British Medical Association (BMA) survey revealed that in ethnic minority doctors, who form nearly one third of the NHS workforce 22, more than 80 per cent believed that their ethnicity had a negative effect on their career advancement 23. In 2004 the Royal College of Psychiatrists accepted that racism existed in the NHS and in their own institution 24.
How does racism manifest itself in medicine?
“Discrimination can appear to be hidden when it is institutionalised, although it is not usually hidden from the person who is subjected to it” 3
Institutional racism is “the collective failure of an organisation to provide an appropriate and professional service to people because of their colour, culture and ethnic origin” 13. Health disparities among patients have been widely linked to racially biased discriminatory health practices 25, 26, 27 but how do structures, processes, and values within an organisation discriminate against those working in the medical profession?
There is considerable evidence to indicate that discriminatory practices against doctors evolved from medical school. For instance racial discrimination has operated at the time when students applied to study medicine 19, 28, through short-listing based on whether applications had Asian or English names 29, 30, and with downgrading of non-English names by computer 31. Discrimination has also been reported during medical school in the US and Canada 11. UK ethnic minority medical students also perform poorly in examinations compared to white students 32, 33 although the lack of evidence of explicit discrimination may suggest the involvement of more subtle communication styles and cultural differences 33.
If the problems at medical school are accountable by racial organisational processes it is not surprising that discriminatory practices persist after qualification (figure 2)
Figure 2: How BME doctors may experience racism 17, 19, 34, 35, 36
Bullying and harassment
More likely to experience bullying and harassment.
Recruitment and career advancement
More likely to be over-represented in junior grades. Reduced promotion and career advancement also seen in relation to academic careers. Underrepresented in senior leadership positions.
Disciplinary hearings
Over-represented at disciplinary hearings with nearly a third of complaints coming form other health professionals.
Disciplinary action and dismissals
Six times more likely to be disciplined e.g. in 2006 two thirds of the 54 doctors struck off in UK had trained outside UK.
Reward systems
Disadvantaged in the allocation of discretionary grants and NHS distinction awards.
What are the consequences of racism in healthcare?
“Racial discrimination damages both those discriminated against and those doing the discriminating” 37
The cost of workplace racism is that it acts as a chronic and acute stressor on the individual with a range of consequences (figure 3):
Figure 3: Consequences of racism on an individual 1, 10, 38, 39
Psychological
Poor well-being. Loss of confidence. Humiliation. Low morale. Gives a sense of thwarted aspirations.
Physiological
Increase blood pressure. Physical illness.
Behavioural
Bad work performance. Require time off work.
“Racial fatigue” characterises the potential emotional and psychological sequelae of feeling isolated in a work environment in which race regularly influences behaviour but is consistently ignored and nobody wants to discuss it (“racial silence”) 40. Racism may be underreported for the same reasons seen with workplace bullying: fear of making matters worse, belief that nothing will be done, concerns regarding confidentiality, fear of victimisation, and concern about being labelled as a troublemaker 41. In addition the individual may fear being regarded as having a “chip on one’s shoulder”.
Organisations may also suffer with disharmony at work, high sickness levels, and resignation 1. In medicine this results in the “double loss” of a speciality losing highly motivated people and gaining those where enthusiasm may be low 19. In addition victims of racial discrimination in healthcare may pursue legal action. In 2003 a surgeon won over £600,000 42 after being denied entry to the specialist registrar. Another surgeon successfully sued the BMA for more than £800,000 for racial discrimination after it failed to support his own claim against the DOH 43. In another case a UK trust paid £2.5m, including legal costs, for wrongful dismissal of a consultant obstetrician who was investigating discrimination 44.
What can be done if you are experiencing racism at work?
In the UK there is protection by legislation. It is unlawful to discriminate against anyone on racial grounds. The Race Relation Act 1976 defined three types of discrimination (direct, indirect, and victimisation) 1, 45,. Following this was the setting up of the Commission for Racial Equality (CRE) in the UK to tackle racism and promote racial equality 45. The Race Relations Act 1976 has now been superseded by the Race Relations (amendment) Act 2000 46 that requires public bodies to eliminate discrimination, promote equal opportunities, and ensure good race relations. However legal processes are stressful and there are some steps you can take before pursuing this route (figure 4).
Figure 4: Steps to take if you are a victim of racial discrimination 1
Talk to colleagues and friends who may have suffered a similar problem because it helps to share a problem and trying to cope on your own can be particularly stressful.
Keep a diary of events of who said what, when, circumstances and any witnesses – this will give a vital record of the nature of the racism.
Find out whether your employer has specific rules about racism at work or a grievance procedure you can use to raise a problem.
If you are in a union contact them to assist you with talking to management or approaching the perpetrator.
In the UK the Commission for Racial Equality is a national body that can help victims of racial discrimination.
Your local Citizens Advice Bureau or Law Centre may be able to help.
You may want to talk to a private law firm that specialises in discrimination issues.
Recommendations and Conclusion
“The law may be just but its implementation is another matter” 47
Despite legislation and procedures, to address racism at work, healthcare organisations are slow in introducing and supporting the policies for race equality 18. Suits come to legal action, not for a lack of policy, but because of not being enforced 48. Practice is not synonymous with policy 19. Reinforcement of policies depends on the degree to which upper management understands discrimination and harassment 48. Although implementation of policies could be successful in combating overt racism this is not so for the covert form.
The covert form of racism, as in institutional racism where organisational processes are “unwittingly” enacted 13, suggests that racism is inevitable. Even people with strong motivations to avoid it are subject to automatic cognitive activation of stereotypes, which can unconsciously influence behaviour, making diversity training courses and non-discrimination policies relatively ineffective 10. Attending to, and encouraging the reporting of, the “softer” aspects of racism may be the key to establishing a true “positively diverse climate” 10. New forms of racism require new approaches (Figure 5):
Figure 5: The STEEP model to approaching subtle racism in organisations 8
Structured Support
Visibly supported by senior management.
Training and Education
Educate people about subtle bias and training to recognise it.
Experience
Frequent and constructive interracial contact to decrease bias, enhance group cohesion, and increase productivity.
Personal Commitment
Individuals must be committed to recognise and combat subtle racism.
The most important part of the solution is education. Teaching on racism should be incorporated into the undergraduate and postgraduate curriculum 19. However of greater significance is recognising our own personal prejudices, at an early stage, so that prejudices we all harbour are challenged within ourselves.
“The hardest attitude to change is the one you don’t know you have” 8
KEY POINTS – RACISM:
Is associated with power and superiority
Has evolved from an overt to a covert form
Is commonplace in healthcare organizations
Is manifested at each stage of a doctors career
Has implications for individuals and organizations
Can be partly dealt with through policies and legislation
Requires a new approach to eradicate the problem
Useful UK online resources
http://www.oneworkplace.co.uk - “One Workplace Equal Rights” aims to tackle racism and promote equal opportunities in the workplace.
http://homepage.ntlworld.com/rajen/RacialEquality - Race Equality Ltd provides phone advice about racism in the medical establishment.
http://www.bidaonline.org.uk - The British International Doctors Association protects and promotes the interests of Ethnic Minority Doctors and Dentists working in the UK.
http://www.equalityhumanrights.com - A new commission working to eliminate discrimination, reduce inequality, protect human rights and build good relations.
COMPETING INTERESTS None Declared AUTHOR DETAILS MINAL MISTRY, BSc, BM, MRCPsych, MSc, Hampshire Partnership NHS Trust, United Kingdom JAVED LATOO, MBBS, DPM, MRCPsych, North East London NHS Foundation Trust, United Kingdom CORRESPONDENCE: Dr MINAL MISTRY, Hampshire Partnership NHS Trust, Melbury Lodge, Winchester, United Kingdom Email: minalmistry@yahoo.co.uk
References
1. TUC. Racism at work - a crime in anyone’s language. Available from: http://www.tuc.org.uk/tuc/rights_racism.cfm
2. Stainback K, Robinson CL, and Tomaskovic-Devey D. Race and workplace integration: a politically mediated process? Am Behav Sci. 2005; 48(9): 1200-1228.
3. Moore LJ. Psychiatric contributions to understanding racism. Transcult Psychiatry. 2000; 37(2): 147-182.
4. Bhopal R. Racism in medicine. Br Med J. 2001; 322: 1503-1504.
5. Bhopal R. Spectre of racism in health and health care: lessons from history and the United States. Br Med J. 1998; 316: 1970-1973.
6. Baker LD. Racism in professional settings: forms of address as clues to power relations. J Appl Behav Sci. 1995; 31(2): 186-201.
7. Encyclopaedia Britannica. Collective unconscious. Available from: http://www.britannica.com/EBchecked/topic/125572/collective-unconscious
8. Dovidio J. The subtlety of racism. Train Dev J. 1993; 47(4): 50-57.
9. Craig-Henderson KM. The automaticity of stereotyping and discrimination. PsycCritiques. 2006; 51(40): abstract.
10. Deitch EA, Barsky A, Butz RM, Chan S, Brief AP and Bradley JC. Subtle yet significant: the existence and impact of everyday racial discrimination in the workplace. Hum Relat. 2003; 56(11): 1299-1324.
11. Beagan BL. ‘Is this worth getting into a big fuss over?’ Everyday racism in medical school. Med Educ. 2003; 37: 852-860.
12. Aluko Y. American Medical Association apologizes for racism in medicine. J Natl Med Assoc. 2008; 100(10): 1246-1247.
13. Macpherson W. The Stephen Lawrence inquiry report. London: Stationary office, 1999.
14. Beishon S, Virdee S and Hagell A. Nursing in a multi-ethnic NHS. London: Policy studies Institute, 1995.
15. Staines R. Is racism a problem in nursing? Nurs Times. 2006; 102(10): 12-13.
16. Duffin C and Parish C. Freedom from racism. Nurs Stand. 2005: 20(4): 22-23.
17. White C. Trusts are making slow progress in promoting race equality, says Healthcare Commission. Br Med J. 2009; 338: b1357.
18. Sheikh A. What’s to be done about racism in medicine? J R Soc Med. 2001; 94(10): 499-500.
19. Coombes R. Mountains to climb. Health Serv J. 2004; 114(5925): 39-46.
20. Chand K. Letter: NHS racism is swept under the carpet. GP. 2004: 33-34.
21. Coker N (ed). Racism in medicine: an agenda for change. London: Kings Fund, 2001.
22. Esmail A. The prejudices of good people. Br Med J. 1994; 328: 1448-1449.
23. Cooke L, Halford S and Leonard P. Racism in the medical profession: the experience of UK graduates, 2006. Available from: http://www.bma.org.uk/employmentandcontracts/equality_diversity/ethnicity/racism.jsp
24. Mamode N. Is the BMA a 21st century organisation? Br Med J. 2004; 329: 161-163.
25. Dennis GC. Racism in medicine: planning for the future. J Natl Med Assoc. 2001; 93(3) Suppl: 1S-5S.
26. Clark PA. Prejudice and the medical profession; a five year update. J Law Med Ethics. 2009: 118-133.
27. Johnstone M-J. Nurses must take a stand against racism in health care. Inter Council Nurses. 2006; 53: 159-160.
28. Mcmanus IC. Factors affecting likelihood of applicants being offered a place in medical schools in the United Kingdom in 1996 and 1997: retrospective study. Br Med J. 1998; 317: 1111-1117.
29. Esmail A and Everington S. Racial Discrimination against doctors from ethnic minorities. Br Med J. 1993; 306: 691-692.
30. Esmail A and Everington S. Letters: Asian doctors are still being discriminated against. Br Med J. 1997; 314: 1619.
31. Lowry S and Macpherson G. A blot on the profession. Br Med J. 1988; 296: 657-658.
32. McManus IC, Richards P, Winder BC and Sproston KA. Final examination performance of medical students from ethnic minorities. Med Educ. 1996; 30(3): 195-200.
33. Wass V, Roberts C, Hoogenboom R, Jones R and Van der Vleuten C. Effect of ethnicity on performance in a final objective structured clinical examination: qualitative and quantitative study. Br Med J. 2003; 326: 800-803.
34. Fox S and Stallworth LE. Racial/ethnic bullying: exploring links between bullying and racism in the US workplace. J Vocat Beh. 2005; 66(3): 438-456.
35. Carr PL, Palepu A, Szalacha L, Caswell C and Inui T. ‘Flying below the radar’: a qualitative study of minority experience and management of discrimination in academic medicine. Med Educ. 2007; 41: 601-609.
36. Pitcher G. NHS human resources staff accused of ignoring racism and bullying of Asian doctors. Available from: http://www.personneltoday.com/articles/2007/08/20/41980/nhs-human-resources-staff-accused-of-ignoring-racism-and-bullying-of-asian-doctors.html
37. Smith R. Deception in research, and racial discrimination in medicine. Br Med J. 1993; 306: 668-669.
38. Forman TA. The social psychological costs of racial segmentation in the workplace: a study of African Americans’ well-being. J Health Soc Beh. 2003; 44(3): 332-352.
39. McKenzie K. Racism and health. Br Med J. 2003 ;326: 65-66.
40. Nunez-Smith M, Curry LA, Bigby JA, Berg D, Krumholz HM and Bradley EH. Impact of race on the professional lives of physicians of African descent. Ann Intern Med. 2007; 146: 45-51.
41. Mistry M and Latoo J (2009). Bullying: a growing workplace menace. Available from: http://www.bjmp.org/content/bjmp-march-2009-volume-2-number-1
42. Newman M. Who’s representing doctors on racism? Hospital Doctor. 11th Oct 2005: p. 9
43. Saleem A. How racism blocked this doctor’s career. Hospital Doctor. 18th Mar 2004: p. 26-27.
44. Symon A. institutional racism and discrimination: are they endemic in the NHS? Br J Midwifery. 2006; 14(6): 366.
45. Dimond Bridgit. Race relations and the law. Br J Midwifery. 2002; 10(9): 580-583.
46. Race relations (amendment) act 2000: New laws for a successful multi-racial Britain. Available from: http://www.homeoffice.gov.uk/documents/cons-2001-race-relations/
47. Dhruev N. Institutional racism: not just skin-deep: how systems sustain racism. Nurs Times. 2002; 98(23): 26-27.
48. Root M P P. The consequences of racial and ethnic origins harassment in the workplace: conceptualisation and assessment. In Barrett K H and George W H (ed). Race, culture, psychology and law. 2005: p. 125.
Ventilator Associated Pneumonia (VAP) is defined as pneumonia occurring in a patient within 48 hours or more after intubation with an endotracheal tube or tracheostomy tube and which was not present before1, 2. Early onset VAP occurs within 48 hours and late onset VAP beyond 48 hours of tracheal intubation.
Incidence
Between 5-15% of hospital in-patients develop infection during admission to ICU3. Patients are 5-10 times more likely to acquire nosocomial infections than patients in the wards4and approximately 86% of hospital associated pneumonia is linked with mechanical ventilation5.
Approximately 10-28% of critical care patients develop VAP6. VAP is also the most common and fatal infection of ICU7,8 and in the United States it affects 9-27% of intubated patients and doubles the risk of mortality as compared with similar patients without VAP9-13.
VAP may account for up to 60% of all Healthcare-Associated Infections14. VAP increases length of ICU stay by 28%16 and each incidence of VAP is estimated to generate an increased cost of £6000- £2200015.
Diagnosis
Despite the high incidence, diagnosis remains challenging because many conditions common to ICU patients like ARDS, sepsis, cardiac failure and lung atelectasis have similar clinical signs. More than 50% of patients diagnosed with VAP do not have the disease whereas upto one-third are not diagnosed17, 18. Unfortunately there is no clearly accepted gold standard for diagnosis of VAP19.
Centres for disease control and prevention (CDC) national healthcare safety network definition for VAP
Radiology signs (2 or more serial chest x-rays with at least one of the following)
New or progressive and persistent infiltrate
Consolidation
Cavitation
Clinical signs
At least one of the following
Fever (temperature > 38 deg C with no other recognised cause)
Leucocytosis > 12000WCC/uL or leucopenia (<4000 WCC /uL)
For adults 70 years or older, altered mental status with no other recognisable cause
and at least 2 of the following
New onset of purulent sputum, or change in character of sputum, or increased respiratory secretions, or increased suctioning requirements
New-onset or worsening cough, or dyspnoea or tachypnoea
Rales or bronchial breath sounds
Worsening gas exchange (eg. O2 desaturations [PaO2/FiO2 ≤ 240], increased O2 requirements, or increased ventilation demand)
Microbiological criteria (optional)
At least one of the following:
Positive growth in blood culture not related to another source of infection
Positive growth in culture of pleural fluid
Positive quantitative culture from bronchoalveolar lavage (≥104 colony forming units/ml) or protected specimen brushing (≥103colony forming units/ml)
5% or more of cells with intracellular bacteria on direct microscopic examination of Gram-stained bronchoalveolar lavage fluid
Histopathological evidence of pneumonia
Histological landmark of VAP is multifocal disease favouring dependant lung segments, often at different stages of development and severity with cultures growing heterogenous microbial flora.20,21
Risk Factors
Mechanical ventilation with Endotracheal intubation including Tracheostomy
Prolonged mechanical ventilation
Advanced age
Pre-existing sinusitis and lung disease
Micro or macroaspiration of oropharyngeal or gastric contents
Malnuitrition and immunosuppression
Obesity
Chronic lung disease
Several factors affect the aetiology of VAP
Time of onset of hospitalisation
Stress induced flora change
Antibiotic induced flora change
Exposure to contamination with nosocomial pathogens
Patient interventions
Pathogenesis:
VAP that occurs within 48 hours after tracheal intubation is usually termed as early onset often resulting from aspiration, which complicates intubation process22. VAP occurring after this period is late onset. Early onset VAP is often due to antibiotic sensitive bacteria (eg oxacillin-sensitive Staphylococcus aureus, Hemophilious influenza and Streptococcus pneumoniae), whereas late onset VAP is frequently caused by antibiotic resistant pathogens (eg.oxacillin-resistant Staphylococcus aureus, Pseudomonas aeruginosa, acinetobacter species and enterobacter species) 23,24,25
The pathogenesis of VAP usually requires that two important processes take place:
Bacterial colonisation of the aero-digestive tract
Aspiration of contaminated secretions into the lower airway26.
Therefore, the strategies to prevent VAP usually focus on reducing the burden of bacterial colonisation in the aero-digestive tract, decreasing the incidence of aspiration or both.
The presence of invasive medical devices is an important contributor to the pathogenesis and development of VAP27. Many patients have nasogastric tubes that predispose them to gastric reflux and increase the potential for aspiration. Endotracheal tubes facilitate bacterial colonisation of the tracheo-bronchial tree and lower airway aspiration of contaminated secretions through mucosal injury, pooling of contaminated secretions above the endotracheal tube cuff and elimination of the cough reflex26. The ventilator circuit and the respiratory-therapy equipment may also contribute to the pathogenesis of VAP if they become contaminated with bacteria, which usually originate in the patient’s secretions26, 28.
Prevention:
The National Institute of Clinical Excellence (NICE) in collaboration with National Patient Safety Agency (NPSA) is examining four technical patient safety solutions for the prevention of VAP and in the process of publishing guidelines. The latest technical patient safety solutions for VAP was published in August 2008 which says
1. Body position-mechanically ventilated and intubated patients should be positioned with their upper body elevated for as long as possible. This may be inappropriate in some patients. eg. spinal injuries. 2. Oral antiseptics e.g. 2% chlorhexidine should be included as part of oral hygiene regimen for all patients who are intubated and ventilated. There is insufficient evidence to recommend any particular antibiotic regimen. 3. Use of kinetic beds - a lack of robust evidence meant the Committee was unable to make recommendations for action on the use of kinetic beds. 4. Care bundles - although the evidence supported the use of elements of care bundles; there was insufficient evidence to recommend a care bundle of any specific design.
The Department of Health published the following ‘High impact intervention’ for ventilated patients in June 2007
Elevation of the head of bed to 35-40 degrees
Sedation holding
Deep Vein Thrombosis prophylaxis
Gastric ulcer prophylaxis
Appropriate humidification of inspired gas
Appropriate tubing management
Suctioning of respiratory secretions (including use of gloves and decontaminating hands before and after the procedure)
Routine oral hygiene as per local policy
In addition the following also may contribute to the prevention of VAP
Prolonged nasal intubation (more than 48hrs) should be avoided because of the association between nosocomial sinusitis and ventilator-associated pneumonia29.
Several investigations have suggested that secretions that pool above the inflated endotracheal tube cuffs may be a source of aspirated material and thus VAP. Endotracheal tubes with separate dorsal lumen above the cuff to suction pooled secretions from the subglottic space are now available. The pressure of the endotracheal tube cuff should be adequate to prevent the leakage of colonised subglottic secretions into the lower airway26,30.
Antibiotic Administration:
Previous administration of antibiotics is an important risk factor for VAP because of the presence of antibiotic-resistant bacteria31. In an attempt to reverse the trend towards increasing rates of antimicrobial resistance among hospital acquired infections, more effective strategies for using antibiotics have been advocated that restrict antibiotic use or offer guidelines for their use 32, 33. Eliminating or reducing the unnecessary use of antibiotics should be the primary goal in reducing antibiotic-resistant nosocomial infections32.
The routine use of prolonged courses of empirical therapy i.e. therapy not supported by results of clinical cultures should be avoided to minimise the subsequent development of antibiotic-resistant infections.
The use of aerosolised antibiotics for the prevention of VAP has been abandoned because of its lack of efficacy and subsequent emergence of antibiotic-resistant infections28.
Similarly the routine use of selective digestive tract decontamination has not gained acceptance in the UK and USA because of its lack of demonstrated effect on mortality, emergence of antibiotic resistant infections and additional toxicity. NICE is currently in consultation for selective decontamination of digestive tract guidelines. The technical patient safety solutions for VAP in adults were published in August 2008.
The Committee examined evidence, which suggested that selective decontamination of the digestive tract (SDD) using topical antibiotics may reduce the incidence of VAP and that SDD regimes that include systemic antibiotics may also reduce mortality. However, Specialist Advisers stated that UK intensive care specialists had particular concerns about the risk of infection with Clostridium difficile and the induction and/or selection of resistant, including multiresistant, microorganisms as a result of SDD. Therefore the Committee recommended further research into SDD in a UK setting.
Use of broad-spectrum antibiotics is also not recommended for the prevention of VAP because of increasing antibiotic resistance among subsequent hospital acquired infections. Targeted antibiotic therapy with appropriate dose of appropriate antibiotic is the sensible thing to do.
Vaccines:
Various vaccination programmes in adults and children have reduced the incidence of pneumonia caused by specific pathogens including H.influenzae type B, Streptococcus Pneumoniae and Influenza virus 34, 35.Vaccinations against these may prevent some hospital acquired infections. Pneumococcal and influenza vaccination must be considered before hospital discharge or included in the discharge planning for all patients at increased risk for subsequent respiratory infections including VAP.
Newer Developments:
There have been new advances in equipment and techniques to help prevention of VAP
Endotracheal and tracheostomy tubes with an extra subglottic port to clear pooled secretions above the endotracheal and tracheostomy tube cuff.
Continuous suctioning of the subglottic secretions.
Endotracheal tubes with specially designed cuffs that do not allow pooled secretions above the cuff to trickle down causing micro-aspiration and ultimately leading to VAP. eg. Endotracheal tubes with microthin polyurethane cuff.
Specially designed closed Tracheal Suctioning Systems (TSS) as compared to open tracheal suctioning systems. However a meta analysis of randomised controlled trial showed that closed suctioning system is not associated with a lower incidence of VAP or mortality as compared to open suctioning36
ACKNOWLEDGEMENTS We thank Dr. S. Parida (Consultant Microbiologist) for her valuable contribution in helping us write this manuscript. COMPETING INTERESTS None Declared AUTHOR DETAILS HARSHAL WAGH, Registrar in Anaesthesia, St Albans City Hospital, United Kingdom DEVARAJA ACHARYA, Consultant in Anaesthesia and Critical Care, St Albans City Hospital, United Kingdom CORRESPONDENCE: DR HARSHAL WAGH, Registrar, Dept of Anaesthesia, Level I, St Julians Ward, St Albans City Hospital, St Albans, UK, AL3 5PN Email: drhdw@hotmail.com
References
Prevention of ventilator-associated pneumonia: consultation NICE, Sept 2007
Eggimann P, Pittet D. Infection control in the ICU. Chest 2001; 120: 2059-93
Weber DJ, Raasch R, Rutala WA. Nosocomial infections in the ICU: the growing importance of antibiotic-resistant pathogens. Chest 1999; 115: 34S-41S
Richards MJ, Edwards JR, Culver DH, Gaynes RP. Nosocomial infections in medical intensive care units in the United States. National Nosocomial infections Surveillance System.Crtic Care Med.1999 May; 27950: 887-92
The prevalence of nosocomial infection in intensive care units in Europe. Results of the European Prevalence of Infection in Intensive Care (EPIC) Study. EPIC International Advisory Committee 1995.
Legras A, Malvy D, Quinioux AI, et al. Nosocomial Infections: prospective survey of incidence in five French intensive care units. Intensive Care Med.1998; 24: 1040-1046.
Urli T, Perone G, Acquarolo A, Zappa S, Antonini B, Ciani A. Surveillance of infections acquired in intensive care: usefulness in clinical practise. J Hosp Infect. 2002; 52: 130-135
Heyland DK, Cook DJ, Griffith L, keenan SP, Brun-Buisson C; Canadian Critical Trials Group. The attributable morbidity and mortality of ventilator-associated pneumonia. Am J Respir Crit Care Med. 1999; 159:1249-1256
Fagon JY, Chastre J, Vuagnat A, Trouillet JL, Novara A, Gilbert C. Nosocomial pneumonia and mortality among patients in intensive care units. JAMA 1996; 275:866-869.
Rello J, Quintana E, Ausina V, et al. Incidence, etiology and outcome of nosocomial pneumonia in mechanically ventilated patients. Chest, 1991; 100:439-444.
Jiménez P, Torres A, Rodriguez-roisin R, et.al. Incidence and etiology of pneumonia acquired during mechanical ventilation. Crit care Med.1998; 17:882-885.
Safdar N, Dezfulian C, Collard HR, Saint S. Clinical and economic consequences of ventilator-associated pneumonia; a systemic review.Crit Care Med.2005; 33:2184-2193
CDC. Guidelines for preventing Health -Care -Associated Pneumonia, 2003. Recommendation of the CDC and the Healthcare Infection Control Practices Advisory Committee. MMWR 2004; 53(No.RR-3)
Nosocomial pneumonia: incidence, morbidity and mortality in the intubated-ventilated patient. Pittett 1994
Nosocomial viral ventilator-associated pneumonia in the intensive care unit: a prospective cohort study. Pfr Vincent 2005
Petersen IS, Aru A, SkØdt V, et al. Evaluation of pneumonia diagnosis in intensive care patients. Scand J infect Dis.1999; 31:299-303
Fagon JY, Chastre J, Hance AJ, Domart Y, Trouillet JL, Gilbert C. Evaluation of clinical judgement in the identification and treatment of nosocomial pneumonia in ventilated patients.Chest.1993; 103:547-553
American Thoracic Society; Infectious diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med.2005; 171:388-416.
Rouby JJ, Martin De Lassale E, Poete P, et al. Nosocomial bronchopneumonia in the critically ill: histologic and bacteriologic aspects. Am Rev Respir Dis.1992; 146:1059-1066.
Fabregas N, Torres A, El-Ebiary M, et al. Histopathologic and microbiologic aspects of ventilator-associated pneumonia. Anaesthesiology.1996; 84:760-761
Pingleton SK, Fagon JY, Leeper KV Jr. Patient selection for clinical investigation of ventilator-associated pneumonia: criteria for evaluating diagnostic techniques. Chest 1990; 97:170-81.
Niederman MS,Craven DE, Fein AM, Schultz DE. Pneumonia in the critically ill hospitalised patient. Chest 1990; 97:170-81.
Kollef MH, Silver P,Murphy DM, Trovillion E. The effect of late-onset ventilator-associated pneumonia in determining patient mortality. Chest 1995; 108:1655-62.
Rello J, ausina V, Ricart M, Castella J, Prats G. Impact of previous antimicrobial therapy on the therapy on the etiology and outcome of ventilator-associated pneumonia. Chest 1993; 104:1230-5.
Craven DE, Steger KA. Epidemiology of nosocomial pneumonia: new perspectives on an old disease. Chest 1995; 108: Suppl: 1S-16S.
Kollef M. Current concepts - the prevention of VAP. NEJM 340; 8:627-634.
Tablan OC, Anderson LJ, Arden NH, Breiman RF, Butler JC, Mcneil MM. Guideline for prevention of nosocomial pneumonia: The Hospital Infection Control Practices Advisory Committee, Centres For Disease Control And Prevention.Infect Control Hosp Epidemiol 1998; 19:304
Rouby JJ, Laurent P, Gosnach M et al. Risk factors and clinical relevance of nosocomial maxillary sinusitis in the critically ill. Am J Respir Crit Care Med 1994; 150: 776-83
Valles J, Artigas A, Rello J et al. Continuous aspiration of subglottic secretions in preventing ventilator associated pneumonia. Ann Intern Med 1995; 122:179-86.
Crouch Brewer S, Wuderink RG, Jones CB, Leeper KV Jr. Ventilator-associated pneumonia due to Pseudomonas aeruginosa. Chest 1996; 109:1019-29
Goldman DA, Weinstein RA, Wenzel RP, et al. Strategies to prevent and control the emergence and spread of antimicrobial-resistant microorganisms in hospitals: a challenge to hospital leadership. JAMA 1996; 275:234-40.
Evans RS, Pestotnik SL, Classen DC, et al. A computer-assisted management programme for antibiotics and other anti-infective agents. N Engl J Med 1998; 338:232-8.
Herceg A.The decline of Haemophilus influenza type b disease in Australia.Commun Dis Intell 1997; 21:173-6.
Gross PA, Hermogenes AW, Sacks HS, Lau J, Levandowski RA. The efficacy of influenza vaccine in elderly persons: a meta analysis and review of literature. Ann Intern Med 1995; 123:518-27.
I I Siempos, K Z Vardakas, M E Falagas. Br J Anaesth 2008; 100(3): 299-306.
Technical patient safety solutions for Ventilator-associated pneumonia in adults. NICE August 2008
Osteoporotic
fracture is common, expensive, and associated with increased morbidity
and mortality. The incidence of osteoporosis fracture annually is greater
than the risk of stroke, breast cancer, and heart attack combined. Bisphosphonates
(BPs) have recently been the subject of clinical controversies because
of the reported incidence of osteonecrosis of jaw (ONJ). Bisphosphonates
as a group of drugs were introduced for the management of various conditions
such as osteoporosis, Paget’s disease, multiple myeloma, and hypercalcemia
of malignancy. This group of drugs has improved the quality of life
in many patients with proven efficacy in limiting pain and skeletal-related
events. The efficacy of BPs as one method to prevent and treat osteoporosis
and avert future fractures, particularly vertebral fractures, is well
documented in large clinical trials. However, despite this evidence,
many patients at risk for osteoporosis are not screened or treated.
The controversy of osteonecrosis of the jaws and bisphosphonates is
a recent and growing problem.
Bisphosphonates:
Bisphosphonates
are fairly safe drugs to be used in the long term. There is a significant
amount of safety data for up to 10 years with alendronate or Fosamax
and up to 7 years with risedronate or Actonel. Every year, an estimated
30 million BP prescriptions are written in the U.S. alone.1
The bisphosphonates alendronate, risedronate, ibandronate, and zoledronic
acid are nitrogen containing compounds that increase bone mineral density
(BMD) by inhibiting osteoclast-mediated bone resorption.2
They have been shown to increase BMD approximately 2–8%, depending
upon the dose and site measured, and have demonstrated efficacy in primary
and secondary prevention of osteoporotic fractures.3–10
Nitrogen-containing
bisphosphonates are used widely for the management of metastatic cancer
in bone (intravenous zoledronic acid or Pamidronate), for the prevention
and treatment of osteoporosis (oral alendronate, risedronate, and ibandronate
and intravenous ibandronate), for the treatment of Paget’s disease
of bone (intravenous Pamidronate and oral alendronate and risedronate),
and for the short-term management of acute hypercalcemia (intravenous
zoledronic acid and Pamidronate).11 Bisphosphonates reduce
the survival and function of osteoclasts, the bone-resorbing cells.
The clinical pharmacology of intravenous (IV) BPs is characterized by
low intestinal absorption but highly selective localization and deposition
in bone. Oral BPs have a bioavailability of less than 5%.12
Once in the blood, BPs disappear very rapidly into the bone.13
After BPs are buried in the skeleton, they are released only when the
bone is destroyed in the course of turnover. In humans, the skeletal
half-lives of various BPs range from 3 months to as long as 10 years.
14
Osteoporosis:
Osteoporosis
is a devastating disease that may lead to significant morbidity and
mortality from resultant fractures. Approximately one in two women and
one in four men over age 50 will have an osteoporosis related fracture
in their remaining lifetime. 15 According to estimated
figures, osteoporosis was responsible for more than 2 million fractures
in US in 2005 15 (including approximately 297,000 hip fractures,
547,000 vertebral fractures, 397,000 wrist fractures , 135,000 pelvic
fractures , 675,000 fractures at other sites). The number of fractures
due to osteoporosis is expected to rise to more than 3 million by 2025.
15
Osteoporotic
fractures are associated with significant morbidity and mortality. Patients
who sustain a fracture are more likely to have lower health-related
quality of life, depression, pain, disability, physical deconditioning
due to inactivity, vertebral deformities with a resultant decrease in
pulmonary function and increase in gastrointestinal complications
(e.g., refractory reflux esophagitis), pressure ulcers, increased likelihood
of nursing home placement, and changes in self-image. 16-23
Hip fractures, which are the most serious complication of osteoporosis,
are associated with significant mortality. 24–27 Up to
38% of patients may die within one year after a hip fracture, and the
risk of death is approximately double that of patients who do not sustain
a hip fracture. 24, 25, 27
The economic
consequences of osteoporosis are enormous. In 1995 in USA, osteoporotic
fractures were responsible for approximately 432,000 hospital admissions,
2.5 million physician’s office visits, and 180,000 nursing home admissions.
15 Health care costs associated with osteoporotic fractures in
2005 were an estimated $19 billion. By 2025, experts predict that these
costs will rise to approximately $25.3 billion. 15 As the
population of the United States continues to age, these costs will likely
increase, with the number of hip fractures and associated costs possibly
tripling by 2040. 15
Oral
bisphosphonate associated osteonecrosis of the jaw:
Osteonecrosis
of the jaws (ONJ) is characterized by the death of bone as a natural
consequence of a wide variety of systemic and local factors compromising
the blood flow of the bone. Clinically it is diagnosed by an area of
exposed bone in the mandible, maxilla, or palate that typically heals
poorly or does not heal over a period of 6 to 8 weeks. The diagnosis
is primarily a clinical one, but imaging studies such as computed tomography
can be helpful. Approximately two thirds of cases involve the mandible
and the rest involve the maxilla. The lesion is painful in many, but
not all, patients, and infection is often present. In one unusual case,
osteonecrosis of the external auditory canal developed in a patient
with myeloma who had received intravenous zoledronic acid and amidronate.28
Predisposing factors for the development of osteonecrosis of the jaw
appear to be dental disease, dental surgery (e.g., tooth extraction),
oral trauma, periodontitis, and poor dental hygiene. The risk factors
for developing ONJ include trauma, female gender, advanced age, edentulous
regions, radiotherapy, chemotherapy, steroid therapy, blood dyscrasias/metastatic
disease, anemia, coagulopathy, surgical dental procedures, alcohol or
tobacco use, prior infection, and bisphosphonate therapy.29-32
Although there have been some reports in the literature about osteonecrosis
caused by steroids, this form is different from ONJ in the sense that
steroid-induced osteonecrosis does not cause bone exposure.
1,
33
ONJ in connection
with bisphosphonate use was first reported in 2003 34, or
5 to 10 years after these drugs were approved in the United States for
their current indications; it was rarely seen before then. Most of the
reported cases (95%) have been associated with zoledronic acid or Pamidronate
given intravenously to control metastatic bone disease. 35, 36,
11 Myeloma and breast cancer are by far the most common cancers
associated with intravenous bisphosphonate use and osteonecrosis of
the jaw.35
Osteonecrosis
of the jaw has developed far less often among patients who have received
oral bisphosphonates at the lower doses used for osteoporosis than among
patients who received the higher doses used for metastatic cancer. Among
several million patients who have received oral treatment for osteoporosis,
fewer than 50 cases of osteonecrosis of the jaw have been reported to
date.35 Moreover, with more than 60,000 patient- years of
exposure to nitrogen- containing bisphosphonates in clinical trials
of treatment for osteoporosis (involving follow-up for as long as 10
years in some patients), osteonecrosis of the jaw was not reported among
the adverse events. 11 The exact incidence of ONJ is unknown.
However, some reports have estimated it to be about 1 in 10,000 for
Intravenous use of BPs. 37 1 in 100,000 patient years
is a reasonable estimate of the incidence of osteonecrosis of the jaw
in patients receiving oral nitrogen-containing bisphosphonates for osteoporosis.
11 The risk of developing ONJ for patients taking alendronate,
the most commonly prescribed oral bisphosphonate, has been estimated
to occur in approximately 0.7 per 100,000 persons per years’ exposure
38; on the other hand, the incidence of ONJ for risedronate and
ibandronate cannot yet be quantified because too few cases have been
reported (12 cases for risedronate and one for ibandronate). 38
The Cartosos
medical claims database study also surveyed 260,000 subjects with osteoporosis,
and found an odds ratio for inflammatory necrosis of the jaw to be 0.65
in oral bisphosphonate users, and that for surgery for a necrotic process
to be 0.86. 39 Both these values are consistent with the
other data suggesting that oral bisphosphonate use does not increase
ONJ risk in osteoporosis patients. These findings are very similar to
those from a case-control study using a claims database, which found
that receiving at least
one oral bisphosphonate prescription was associated with an odds ratio for
jaw surgery of 0.91. 40 As per one consensus panel, there
have been 33 cases [reported] in the literature as of January 2007 --
out of the 33 million patients who have been treated worldwide with
an oral bisphosphonate -- which translates into approximately 200 million
prescriptions written. 41 In addition, there has been spontaneous
reporting in 1 of 100,000 patient-years for all of the approved bisphosphonates.
41 The fact that the majority of reported cases of osteonecrosis
of the jaw are associated with the use of high-dose intravenous bisphosphonates
for metastatic bone disease suggests that the dose, duration of treatment,
and route of administration, as well as coexisting conditions, concomitant
treatments (glucocorticoids or immunosuppressive agents), and dental
health, could all be related to the incidence of this complication.
11
Prevention
and management of bisphosphonate-associated osteonecrosis of the jaw:
Published
recommendations are based upon expert experience from a variety of sources.
35,36,1, 42-–45 As yet, there have been no randomized, controlled
trials that have evaluated strategies to prevent or manage ONJ in individuals
receiving long-term high-dose bisphosphonate therapy. Before initiating
BP therapy, all medical and dental practitioners are encouraged to follow
these guidelines:
All patients
should undergo a routine dental exam to rule out any dental source of
infection.
All medical practitioners
also should perform a baseline oral exam.
Invasive dental
or/and oral surgical procedures should be completed before initiating
therapy.
Practice preventive
dentistry, involving procedures such as oral prophylaxis, dental restorations,
and endodontic therapy, and check dentures for irritational foci.
Schedule routine
follow-up every 3 months to check for any signs of developing ONJ.
The risks associated
with oral surgical procedures such as dental implants, extractions,
and extensive periodontal surgeries must be discussed with the patient
and weighted against the benefits.
The following
recommendations are made by the American Association of Oral and Maxillofacial
Surgeons for management of patients on BP therapy and patients with
proven ONJ. 46
Management
of patients with proven ONJ based on staging of the condition:
Stage 1: Exposed/necrotic bone in patients who are asymptomatic and have no
evidence of infection
Stage 2: Exposed/necrotic bone in patients with pain and clinical evidence
of infection.
Stage 3: Exposed/necrotic bone in patients with pain, infection, and 1 or
more of the following: Pathologic fracture, extra oral fistula, or osteolysis
extending to the inferior border.
Treatment of patients with established ONJ:
Patients with stage 1 ONJ: Conservative management with oral rinse such as 0.12%
chlorhexidine.
Patients
with stage 2 ONJ: Manage with antibiotics and antimicrobial oral rinses.
Patients
with stage 3 ONJ: Surgical debridement/ resection in combination with
antibiotic therapy.
Extraction
of symptomatic teeth can be performed without any additional risks of
worsening the condition.
General
recommendations:
As with
all dental patients, routine dental examinations are recommended. A
comprehensive oral evaluation should be carried out of all patients
about to begin therapy with oral bisphosphonates (or as soon as possible
after beginning therapy). The dentist should inform the patient taking
oral bisphosphonates that there is a very low risk (estimated at 0.7
cases per 100,000 person-years’ exposure) of developing ONJ; there
are ways to minimize the risk, but not to eliminate the already low
risk; the consensus is that good oral hygiene along with regular dental
care is the best way to lower risk; there are no diagnostic techniques
to identify those at increased risk of developing ONJ.
Before undergoing
any invasive procedure that involves manipulation of the bone the patient
should understand that at this time, the risk of developing osteonecrosis
of the jaw is considered very small, and that the vast majority of patients
taking an oral bisphosphonate do not develop any oral complications.
(Dental management of patients receiving oral bisphosphonate therapy:
Expert panel recommendations)
Based on
the currently available information, National Osteoporosis Foundation
believes that in the vast majority of patient who are receiving them,
the benefits of oral bisphosphonate medications outweigh the potential
risk of ONJ. Patients for whom bisphosphonates are appropriate would
be at higher risk of fractures without treatment, and fractures are
the source of significant pain and disability that impact on function
and quality of life. If a patient receiving bisphosphonates has planned
dental surgery that involves the bone, a drug holiday beginning shortly
before the procedure and lasting until there is local healing could
be considered, although there is as yet no clinical evidence that this
will affect the incidence or severity of ONJ. (Osteonecrosis of the
Jaw (ONJ) June 14, 2006 / Reviewed and approved by the Science and Research
Committee of the NOF Board of Trustees March 3, 2007).
Conclusion:
There is
a need to clearly delineate the incidence of ONJ in osteoporosis patients
treated with oral bisphosphonates, and in appropriate control populations.
Based on current evidence, the risk of ONJ in osteoporosis patients
taking oral BPs appears to be comparable to that in the general population.
With the likely prevalence sitting at approximately 1 per 100,000 patient-years,
it is quite clear that this is no different from that in the general
population, since these problems can certainly occur in the absence
of bisphosphonate use. The documented benefits of using bisphosphonates
for established indications clearly outweigh whatever small risk of
osteonecrosis of the jaw might be incurred. 11, 47 Even if
the number of cases of ONJ in patients taking oral bisphosphonates are
still rare compared to the total exposure, primary care physicians treating
bone diseases with bisphosphonates need to be aware there is a small
risk their patients may develop this new complication, allowing for
prophylaxis, early diagnosis and prevention of potential consequences.
The benefits and risks of bisphosphonate therapy should be individually
discussed and, when necessary and possible, alternative therapy for
postmenopausal osteoporosis should be considered.
It is important
to understand that, based on the information currently available; the
risk for developing BON is much higher for cancer patients on intra
venous bisphosphonate therapy than the risk for patients on oral bisphosphonate
therapy. Therefore, there are different recommendations for dental management
of these patients.
In conclusion,
the risk of ONJ is extraordinarily low. The risk of being in a fatal
car accident is 10-15 times as high as the risk of ONJ from taking an
oral bisphosphonate. 41
Competeing Interests:
Serves as a speaker for Eisai Inc. and Pfizer Inc. for the 2008 ARICEPT LTC DELTA
2 (Dementia Education Leadership Training in Alzheimer's) Promotional
Education Program AUTHOR DETAILS
NASSEER A. MASOODI, MD,
FACP, CMD. Assistant Professor Clinical Sciences, Florida State University
College of Medicine, Tallahassee, FL-USA; Courtesy Assistant Professor
Geriatrics, University of Florida College of Medicine, Gainesville,
FL-USA; Medical Director Health Services, ACV Inc, Dowling Park, FL-USA
CORRESSPONDENCE: PO BOX: 4346, Dowling Park, FL-32064, USA.
Email: haadin@yahoo.com
References
Gutta R, Louis
P. Bisphosphonates and osteonecrosis of the jaws: Science and rationale.
Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007;104:186-93
NIH Consensus
Conference. Optimal calcium intake. NIH Consensus Development Panel
on Optimal Calcium Intake. JAMA. 1994; 272:1942–8.
Cummings SR,
Black DM, Thompson DE et al. Effect of alendronate on risk of fracture
in women with low bone density but without vertebral fractures: results
from the Fracture Intervention Trial. JAMA. 1998; 280:2077–82.
Black DM, Cummings
SR, Karpf DB et al. Randomized trial of effect of alendronate on risk
of fracture in women with existing vertebral fractures. Lancet. 1996;
348:1535–41.
Harris ST, Watts
NB, Genant HK et al. Effects of risedronate treatment on vertebral and
nonvertebral fractures in women with postmenopausal osteoporosis: a
randomized controlled trial. JAMA. 1999; 282:1344–52.
Reginster J,
Minne HW, Sorensen OH et al. Randomized trial of the effects of risedronate
on vertebral fractures in women with established postmenopausal osteoporosis.
Osteoporos Int. 2000; 11:83–91.
McClung MR, Geusens
P, Miller PD et al. Effect of risedronate on the risk of hip fracture
in elderly women. N Engl J Med. 2001; 344:333–40.
Chesnut IC, Skag
A, Christiansen C et al. Effects of oral ibandronate administered daily
or intermittently on fracture risk in postmenopausal osteoporosis. J
Bone Miner Res. 2004; 19:1241–9.
Lyles KW, Colón-Emeric
CS, Magaziner JS et al. Zoledronic acid and clinical fractures and mortality
after hip fracture. N Engl J Med. 2007; 357:1799–809.
Black DM, Delmas
PD, Eastell R et al. Once-yearly zoledronic acid for treatment of postmenopausal
osteoporosis. N Engl J Med. 2007; 356:1809–22.
Bilezikian JP.
Osteonecrosis of the Jaw-Do Bisphosphonates pose a risk? N Engl J Med.
2006; 355:2278–81.
Conte P, Guarneri
V. Safety of intravenous and oral bisphosphonates and compliance with
dosing regimens. Oncologist 2004; 9(Suppl 4):28-37.
Bisaz S, Jung
A, Fleisch H. Uptake by bone of pyrophosphate, diphosphonates and their
technetium derivatives. Clin Sci Mol Med 1978; 54:265-72.
Kasting GB,
Francis MD. Retention of etidronate in human, dog, and rat. J Bone Miner
Res 1992; 7:513-22.
National Osteoporosis
Foundation. Physician’s guide to prevention and treatment of osteoporosis. www.nof.
org/physguide/index.htm (accessed Nov. 2008)
Lips P, Cooper
C, Agnusdei D et al. Quality of life in patients with vertebral fractures:
validation of the Quality of Life Questionnaire of the European Foundation
for Osteoporosis (QUALEFFO). Osteoporos Int. 1999; 10:150–60.
Gold DT, Shipp
KM, Lyles KW. Managing patients with complications of osteoporosis.
Endocrinol Metab Clin North Am. 1998; 27:485–96.
Hallberg I,
Rosenqvist AM, Kartous L et al. Health related quality of life after
osteoporotic fractures. Osteoporos Int. 2004; 15:834–41
Nevitt MC, Ettinger
B, Black DM et al. The association of radiographically detected vertebral
fractures with back pain and function: a prospective study. Ann Intern
Med. 1998; 128:793–800.
Fink HA, Ensrud
KE, Nelson DB et al. Disability after clinical fracture in postmenopausal
women with low bone density: the Fracture Intervention Trial (FIT).
Osteoporos Int. 2003; 14:69–76.
Ensrud KE, Thompson
DE, Cauley JA et al. Prevalent vertebral deformities predict mortality
and hospitalization in older women with low bone mass. J Am Geriatr
Soc. 2000; 48:241–9.
Margolis DJ,
Knauss J, Bilker W et al. Medical conditions as risk factors for pressure
ulcers in an outpatient setting. Age Ageing. 2003; 32:259–64.
Melton LJ 3rd.
Adverse outcomes of osteoporotic fractures in the general population.
J Bone Miner Res. 2003; 18:1139–41.
Empana JP, Dargent-Molina
P, Breart G. Effect of hip fracture on mortality in elderly women: the
EPIDOS prospective study. J Am Geriatr Soc. 2004; 52:685– 90.
Center JR, Nguyen
TV, Schneider D et al. Mortality after all major types of osteoporotic
fracture in men and women: an observational study. Lancet. 1999; 353:878–82.
Cauley JA, Thompson
DE, Ensrud KC et al. Risk of mortality following clinical fractures.
Osteoporos Int. 2000; 11:556–61.
Keene GS, Parker
MJ, Pryor GA. Mortality and morbidity after hip fractures. BMJ. 1993;
307:1248–50.
Hoff AO, Toth
B, Altundag K, et al. Osteonecrosis of the jaw in patients receiving
intravenous bisphosphonate therapy. J Bone Miner Res 2005; 20: Suppl
1:1218. Abstract
Bouquot JE,
McMahon RE. Neuropathic pain in maxillofacial osteonecrosis. J Oral
Maxillofac Surg 2000; 58:1003-20.
Assouline-Dayan
Y, Chang C, Greenspan A, Shoenfeld Y, Gershwin ME. Pathogenesis and
natural history of osteonecrosis. Semin Arthritis Rheum 2002; 32:94-124.
Gruppo R, Glueck
CJ, McMahon RE, Bouquot J, Rabinovich BA, Becker A, et al. The pathophysiology
of alveolar osteonecrosis of the jaw: anticardiolipin antibodies, thrombophilia,
and hypofibrinolysis. J Lab Clan Med 1996;127:481-8.
Damato K, Gralow
J, Hoff A, Huryn J, Marx RE, Ruggiero S, et al. Available at: http://www.fda.gov/ohrms/dockets/ac/05/briefing/ 2005-4095B2_02_12-Novartis-Zometa-App-11.pdf.
(Accessed Oct, 2008).
Zigic TM, Marcous
C, Hungerford DS, Dansereau JV, Stevens MB. Corticosteroid therapy associated
with ischemic necrosis of bone in systemic lupus erythematosis. Am J
Med 1985;79:596.
Marx RE. Pamidronate
(Aredia) and zoledronate (Zometa) induced avascular necrosis of the
jaws: a growing epidemic. J Oral Maxillofac Surg 2003;61(9):1115–7.
Woo SB, Hellstein
JW, Kalmar JR. Bisphosphonates and osteonecrosis of the jaws. Ann Intern
Med 2006; 144:753-6. [Erratum, Ann Intern Med 2006; 145:235.]
Marx RE, Sawatari
Y, Fortin M, Broumand V. Bisphosphonate-induced exposed bone (osteonecrosis/osteopetrosis)
of the jaws: risk factors, recognition, prevention, and treatment. J
Oral Maxillofac Surg 2005; 63:1567-75.
Novartis Pharmaceuticals
Co. Updated safety: possible relationship of Aredia (Pamidronate disodium)
and/or Zometa (zoledronic acid) with osteonecrosis of the jaw [letter
to health care professionals]. Ottawa: Health Canada; Nov 2004. Available
at: www.hc-sc.gc.ca/ hpfb-dgpsa/tpd-dpt/aredia_zometa_hpc_e.html.
(Accessed Nov, 2008).
American Dental
Association Council on Scientific Affairs: Expert panel recommendations:
Dental management of patients receiving oral bisphosphonate therapy.
J Am Dental Assoc 2006, 137:1144-1150
Cartosos VM,
Zhu S, Zavras AI. Bisphosphonate use and the risk of adverse jaw outcomes.
JADA 2008; 139: 23-30.
Pazianas M,
Blumentals WA, Miller PD. Lack of association between oral bisphosphonates
and osteonecrosis using jaw surgery as a surrogate marker. Osteoporos
Int 2008;19: 773-779.
Postmenopausal
Osteoporosis: Putting the Risk for Osteonecrosis of the Jaw Into Perspective
Authors: Stuart L. Silverman, MD, FACR, FACP; Mone Zaidi, MD, PhD, FRCP;
E. Michael Lewiecki, MD, FACP; Regina Landesberg, DMD, PhD (Medscape
Online CME, accessed on April 23, 2007).
Migliorati CA,
Siegel MA, Elting LS. Bisphosphonate-associated osteonecrosis: a long-term
complication of bisphosphonate treatment. Lancet Oncol 2006;7(6):508–14
Expert panel
recommendations for the prevention, diagnosis, and treatment of osteonecrosis
of the jaws. LDA J 2005; 64(3):21–4.
Melo MD, Obeid
G. Osteonecrosis of the jaws in patients with a history of receiving
bisphosphonate therapy: strategies for prevention and early recognition.
J Am Dent Assoc 2005; 136(12):1675–81.
Hellstein JW,
Marek CL. Bisphosphonate induced osteochemonecrosis of the jaws: an
ounce of prevention may be worth a pound of cure. Spec Care Dentist
2006; 26(1):8–12.
AAOMS Position
Paper on Bisphosphonate-Related Osteonecrosisof the Jaws. Available
at: http://aaoms.org/docs/position_papers/osteonecrosis.pdf. (Accessed
October, 2008).
ASHP Therapeutic
Position Statement on the Prevention and Treatment of Osteoporosis in
Adults http://www.ashp.org/DocLibrary/BestPractices/TPS_Osteo.aspx (Accessed Nov 25, 2008)
Adjuvant
modes of breast cancer therapy following surgical intervention mainly
revolve round radiation therapy, chemotherapy, or hormone therapy designed
to eliminate residual cancer cells. The increase in the incidence of
breast cancer with age has sharply focused attention on the link between
incidence and progression. It follows from this that approaches to successful
treatment and patient management would converge on hormonal status as
a beneficial mode of targeted therapy. A number of growth factors, besides
the steroid hormones oestrogen and progesterone, are closely involved
in the growth and metastatic spread of breast cancer. Recent years have
seen intensive studies of the mechanisms of function of growth factors
and the pathways by which they stimulate the growth of cancer cells.
These studies have led the way to the targeting growth factor function
as a means of controlling breast cancer development and secondary spread.
The growth
factor receptors are transmembrane proteins. The binding of growth factors
to the external domain activates these receptors which have tyrosine
kinase activity. This activation therefore leads to the phosphorylation
of signalling proteins down stream in the signalling cascade. This in
turn leads to the expression of genes associated with cell proliferation
and often also to the inhibition of apoptotic loss of cells.
Growth
factors, growth factor receptors and tumour growth and progression
Growth factors
and their receptor play a major part in normal growth and differentiation
and also in tumour development and progression. Growth factors promote
proliferation and induce cancer invasion. Certain growth factors, e.g.
the insulin-like growth factor (IGF) might promote tumour growth by
inhibiting apoptotic loss of cells1. Mutations or over-expression
of growth factor receptors is associated with aggressive cancers and
poor prognosis for patients. Growth factor receptor genes are amplified
in a number of human cancers and this is reflected in the expression
of the respective receptor proteins in the cancers. Growth factor receptors
are transmembrane tyrosine kinase proteins and transduce the signals
imparted by the binding of the growth factors to their specific receptors;
this signal transmission ultimately results in the induction of cell
proliferation.
Among growth
factors of note in the context of this editorial are the epidermal growth
factor (EGF) and the Heregulins constituting a family of EGF related
growth factors. There are several isoforms of Heregulin generated by
alternative RNA splicing of the heregulin gene; these isoforms bind
to their receptors with different degree of affinity. The epidermal
growth factor receptor (EGFr) family (also often referred to as the
erb family) includes HER1 (human epidermal growth factor receptor 1
also called EGFr), HER2, HER3
and HER4. These receptor proteins significantly resemble one another
in aminoacid sequence2. Heregulin can bind HER3 and HER4
receptors but the ligand for HER2 has not been identified3
and so HER2 is often described as an orphan receptor. The receptor protein
consists of an external domain that binds growth factors, a transmembrane
domain and an intracellular domain which possesses tyrosine phosphorylation
sites4. Growth factors and their paralogues bind to these
receptors and induce receptor oligomerisation. This activates the cytoplasmic
kinase domains, which phosphorylate and activate target proteins that
induce the expression of genes responsive to the growth factors. The
binding and activation of the receptors is a highly specific process,
but often more than one receptor might be involved in the signalling
process. In this event heterodimerisation would occur between different
receptors; this seems to enhance the affinity of ligand binding5.
This process of engagement of co-receptors to enhance growth factor
signalling has been described as cross-talk between the receptors. HER2
is known to be involved in cross-talk with EGFr, HER3 as well as HER4.
So HER2 seems to occupy a pre-eminent position in the signalling cascade,
but recently it has been suggested that HER3 might also be a prominent
participant6.
The EGFr
family of receptors have been intensively investigated for their potential
relationship to cancer progression and prognosis and as a potential
route for treatment and patient management. Approximately 25% of breast
cancers show HER2 gene amplification and this correlates aggressive
behaviour and poor prognosis7-10. However, on the positive
side the presence of HER2 receptors has provided a new treatment modality
for many patients.
Monoclonal
antibodies (Herceptin) have been raised against the external domain
of HER2. Herceptin has provided a highly successful mode of treatment
for metastatic breast cancer showing high HER2 expression and HER2 gene
amplification11-13. Blocking receptor function with Herceptin
inhibits tumour growth and possibly also microvascular density associated
with tumours and vascular permeability. Furthermore, Herceptin treatment
appears to reduce VEGF (vascular endothelial growth factor) expression,
tumour associated microvascular density and cell proliferation in breast
cancers xenografted into mice14. Also in murine tumour models,
Herceptin reduces the number of circulating cancer cells even under
circumstances where the tumour is resistant to Herceptin treatment15,
which could be a manifestation of its effects on the vasculature independently
of its inhibition of HER2 signalling.
HER2
expression and tamoxifen resistance
Breast cancer
growth is influenced by the sex steroid hormones oestrogen and progesterone
and growth factors such as EGF and HER2 ligands. Patients with tumours
that are oestrogen receptor (ER) positive are treated with tamoxifen.
The latter binds ER and competitively blocks oestrogen signals. In the
context of the deployment of Herceptin to treat HER2+ patients, it has
emerged that tumours over-expressing HER2 are resistant to tamoxifen.
These patients could benefit from anti-oestrogen therapy combined with
blockade of HER2 signalling16. A randomized trial has indicated
that post-menopausal patients with advanced breast cancer can benefit
significantly from this combination therapy17.
Among other
factors that might confer tamoxifen resistance is AIB1, the steroid
receptor co-activator, which is often amplified in breast cancers. In
vitro studies with breast cancer cells and in vivo investigations of
murine tumours have suggested the involvement of AIB1 in tamoxifen resistance18
and in HER2 signalling19. In primary breast cancer also AIB1
has been linked with tamoxifen resistance20, 21. A second
factor deserving discussion in this context is the possibility that
EGFr and HER2 signalling systems might interact and contribute in this
way to resistance to hormonal therapy. As mentioned elsewhere in this
review, EGFr does recruit HER2 as a co-receptor in signal transduction.
This is of some significance for patients with ER-negative tumours.
For, we showed some years ago that a proportion of ER-ve tumours tended
to be EGFr+ve22-24. So this would suggest the possibility
that patients with ER-ve/EGFr+ tumours could conceivably benefit from
Herceptin treatment (see below).
Another
possible means by which tamoxifen resistance might arise has been suggested
by the finding that tamoxifen and Fulvestrant, also an anti-oestrogen,
appear to be able to induce breast cancer cell invasion in the absence
of E-cadherin25. Cadherins are transmembrane proteins, which
have considerable influence on cancer invasion because they alter intercellular
and cell-substratum adhesion. E-cadherin is regarded as a suppressor
of invasion and growth of carcinomas as the loss or mutation of E-cadherin
leads to the acquisition of invasiveness. Borley et al.25
showed that both tamoxifen and Fulvestrant induced invasion in E-cadherin
deficient MCF7 breast cancer cells, but this did not occur after oestrogen
depletion. These findings add a new dimension to tamoxifen resistance
as potentially being mediated by recurrence resulting from induced invasive
ability.
HER2
expression and adjuvant chemotherapy
It has been
recognised of late that combination of Herceptin with chemotherapy might
yield considerable benefits in terms of reduction of recurrence and
mortality. So HER2 expression has come into the reckoning when considering
the use of adjuvant chemotherapy. Combining Herceptin with either an
anthracycline plus cyclophosphamide or with Paclitaxel, as first-line
therapy for metastatic breast cancer over expressing the HER2 receptor,
has provided significant benefits in terms of objective response, duration
of response and survival as compared with chemotherapy alone. Furthermore,
the benefits were related to the degree of HER2 over-expression26.
A review of 35 clinical trials has indicated that patients with HER2+
cancers might benefit more from anthracycline-based and taxane-based
adjuvant chemotherapy than those with HER2-negative cancers27.
Indeed, anti-HER2 antibody combined with chemotherapy is superior to
HER2 anitibody and anti-oestrogen combination17. The benefits
of adjuvant chemotherapy with anthracyclines to patients with HER2 over-expressing
tumours seem to be beyond reasonable doubt. Gennari et al.28
have provided a combined analysis of eight studies. HER2+ patients on
anthracyclines had superior disease-free as well as overall survival
in comparison with patients on non-anthracycline regimen. No such benefits
emerged for HER2-ve patients, suggesting that one can exclude these
patients from anthracycline adjuvant therapy. Also being investigated
is potential synergy between antibodies against other growth factor
receptors and anti-HER2 antibodies.
Attempts
are also currently in progress to test the efficacy of conjugates of
anti-HER2 antibodies with cytotoxic drugs to achieve targeted delivery
of the cytotoxic agents. Laboratory studies are underway with Herceptin-platinum(II)
complexes29 and Herceptin-microtubule-depolymerising agents30.
Contraindications
of Herceptin regimen
The toxic
side-effects of the administration of monoclonal antibodies were recognised
some years ago. Cardiotoxicity, pulmonary toxicity and infusion-related
problems such as anaphylaxis occur, albeit infrequently with monoclonal
antibody therapies 31, 32. These toxicities have been described
with Herceptin treatment, more so in patients on anthracycline and cyclophosphamide
combined with Herceptin26. Cardiotoxicity could occur in
some patients when Herceptin is administered with anthracyclines33,
34. Herceptin itself can be cardiotoxic in patients receiving
concurrent or prior anthracyclines35, 36. Cardiotoxicity
is not due to structural abnormalities but Herceptin might cause myocardial
dysfunction. The toxicity of anthracyclines and Herceptin could be brought
about by different routes37. As Dinh et al.38
have emphasised, many questions relating to Herceptin treatment still
remain unanswered, e.g. optimising treatment, and combination with conventional
chemotherapeutic agents, among others. Even with these caveats Herceptin
may be regarded as a most efficacious agent in the treatment of HER2+
breast cancers.
EGFr
inhibitor Lapatinib (Tykerb) in breast cancer treatment
As stated
earlier, EGFr is over expressed in a proportion of breast cancers that
are ER-negative. EGFr expression also correlates with the expression
of metastasis promoting genes. Further, in the light of the function
of EGFr in conjunction with HER2, it would be of considerable clinical
benefit to test the effects of EGFr inhibitors in breast cancer treatment.
Lapatinib is a powerful dual inhibitor of EGFr and HER2 with marked
pharmacological potential.
Lapatinib
is a protein kinase inhibitor (a 4-anilinoquinazoline derivative), which
inhibits growth factor signalling by binding to the ATP-binding pocket
of both EGFr and HER2 receptor proteins and so prevents autophosphorylation
of the receptor and inhibits the signalling cascade leading to the suppression
of the growth of tumours, including advanced or metastatic breast cancers
resistant to Herceptin39. Objective responses have been achieved
in 28% of patients with untreated HER2-positive tumours40.
Clinical trials have provided promising results; Lapatinib is clinically
very effective especially in advanced or metastatic breast cancer and
patients with brain metastases. A phase III trial assessing the efficacy
of combination of Lapatinib with Capecitabine, which is converted to
5-Flurouracil and inhibits DNA synthesis, seems to suggest a significant
slowing down of disease progression by the combination as compared with
capecitabine alone41. The efficacy of combining Lapatinib
with other conventional chemotherapeutic agents is being evaluated.
First-line Paclitaxel-Lapatinib combination gave significant benefits
to HER-2-positive patients42.
Lapatinib
could be functioning synergistically with HER2 inhibitors, for its effect
on prominently EGFr over-expressing cancers, such as colorectal cancer
or squamous cell carcinoma of the head and neck, is said to be unexceptional
and moderate43. According to Press et al.44 the
benefits of Lapatinib appear to be restricted to patients with HER2
over-expressing cancers. Lapatinib has no cardiac toxicity but does
produce other toxic effects45. However, its toxicity whilst
administered in combination with other anticancer agents has not been
appraised.
Avastin
in breast cancer treatment
A different
route to control of tumour growth and metastatic spread has been afforded
by inhibitors that target the microvasculature associated with tumours.
Tumours induce the formation of neovascularisation so that tumour cells
can access the vascular system and become disseminated to form distant
metastases. The neovasculature is induced by VEGF, which transduces
its effects by binding specifically to its receptors VEGFr (see46).
A strategy similar to that adopted with EGF family growth factors has
been used to target VEGFr, inhibit its function and inhibit the signalling
by VEGF. Avastin (Bevacizumab), a humanised monoclonal anti-VEGFr antibody,
is such an inhibitor. Avastin with Paclitaxel chemotherapy has been
found to enhance progression-free survival and improve response rate
in patients with advanced breast cancer47. However, on account
of possible side effects, there is some reluctance to the use of Avastin.
It has been approved in Europe for first line treatment of women with
metastatic breast cancer, but has not been approved for use by the National
Institute for Health and Clinical Excellence UK48.
ACKNOWLEDGEMENTS
The author
thanks Professor Leif Bergkvist of the Department of Surgery and Centre
for Clinical Research of Uppsala University Central Hospital at Västerås,
and Dr M.S. Lakshmi for reading the manuscript and making helpful suggestions,
and Professor Bayan Sharif and Professor Satnam Dlay for research and
literary facilities
COMPETING
INTERESTS
None Declared AUTHOR
DETAILS
GAJANAN
V SHERBET, The Institute for Molecular Medicine, Huntington Beach CA,
USA and School of Electrical, Electronic and Computer Engineering, University
of Newcastle upon Tyne UK
CORRESPONDENCE:
GAJANAN V SHERBET, School of Electrical, Electronic and Computer Engineering,
University of Newcastle upon Tyne, Merz Court, Newcastle upon Tyne,
NE1 7RU, U.K.
Email:
gajanan.sherbet@ncl.ac.uk
References
Werner H, Le
Roith D. The insulin-like growth factor-I receptor signaling pathways
are important for tumorigenesis and inhibition of apoptosis., Critical
Reviews Oncol 1997; 8: 71-92.
Schlessinger
J, Ullrich A. Growth factor signalling by receptor tyrosine kinases.
Neuron 1992; 9: 383-391
Tzahar E, Levkowitz
G, Karunagaran D, et al. ErbB-3 and ErbB-4 function as respective low
and high affinity receptors of all Neu differentiation factor Heregulin
isoforms. J Biol Chem 1994; 269: 25226-25233.
Ullrich A, Schlessinger
J. Signal transduction by receptors with tyrosine kinase activity. Cell
1990; 61: 203-212.
Karunagaran D,
Tzahar E, Beerli RR, et al. ErbB-2 is a common auxiliary subunit of
NDF and EGF receptors: Implications for breast cancer. EMBO J 1996;
15: 254-264.
Lee-Hoeflich
ST, Crocker L, Yao E, et al. A central role for HER3 in HER2-amplified
breast cancer: implications for targeted therapy. Cancer Res 2008; 68:
5878-5887.
Slamon DJ, Clark
G, Wong S et al. Human breast cancer: correlation of relapse and survival
with amplification of the HER-2/neu oncogene. Science 1987; 235: 177-181.
Slamon DJ, Godolphin
W, Jones L, et al. Studies of the HER-2/neu proto-oncogene in human
breast and ovarian cancer. Science 1989; 244: 707-711.
Venter D, Kumar
S, Tuzi N, et al. Over expression of the c-erbB-2 oncoprotein in human
breast carcinomas: immunohistochemical assessment correlates with gene
amplification. Lancet 1987; 2: 69-72.
Natali P, Nicotra
M, Brigotti A, et al. Expression of the p185 encoded by HER2 oncogene
in normal and transformed human tissues. Int J Cancer 1990; 45: 457-461.
Cobleigh MA,
Vogel CL, Tripathy D, et al. Multinational study of the efficacy and
safety of humanized anti-HER2 monoclonal antibody in women who have
HER2-overexpressing metastatic breast cancer that has progressed after
chemotherapy for metastatic disease. J Clin Oncol 1999; 17: 2639-2648.
Vogel CL, Cobleigh
MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single
agent in first-line treatment of HER2-overexpressing metastatic breast
cancer. J Clin Oncol 2002; 20: 719-726.
Shepard HM,
Jin P, Slamon DJ, et al. Herceptin Handbook Exp Pharmacol 2008;
181: 183-219.
Le XF, Mao WQ,
Lu CH, et al. Specific blockade of VEGF and HER2 pathways results in
greater growth inhibition of breast cancer xenografts that over express
HER2. Cell Cycle 2008; 7: 3747-3758.
Barok M, Balazs
M, Nagy P, et al. Trastuzumab decreases the number of circulating and
disseminated tumor cells despite trastuzumab resistance of the primary
tumor. Can Lett 2008; 260: 198-208.
Rastelli F,
Crispino S. Factors predictive of response to hormone therapy in breast
cancer. Tumori 2008; 94: 370-383.
Prat A, Baselga
J. The role of hormonal therapy in the management of hormonal-receptor-positive
breast cancer with co-expression of HER2. Nature Clin Pratice Oncol
2008; 5: 531-542.
Su QB, Hu SY,
Gao HD, et al. Role of AIB1 for Tamoxifen resistance in estrogen receptor-positive
breast cancer cells. Oncology 2008; 75: 159-168.
Fereshteh MP,
Tilli MT, Kim SE, et al. The nuclear receptor coactivator amplified
in breast cancer-1 is required for neu (ErbB2/HER2) activation, signaling,
and mammary tumorigenesis in mice. Cancer Res 2008; 68: 3697-3706.
Kirkegaard T,
McGlynn LM, Campbell FM, et al. Amplified in breast cancer 1 in human
epidermal growth factor receptor-positive tumors of tamoxifen-treated
breast cancer patients. Clin Cancer Res 2007; 13: 1405-1411.
Dihge L, Bendahl
PO, Grabau D, et al. Epidermal growth factor receptor (EGFR) and the
estrogen receptor modulator amplified in breast cancer (AIB1) for predicting
clinical outcome after adjuvant tamoxifen in breast cancer. Breast Cancer
Res Treat 2008; 109: 255-262.
Sainsbury JRC,
Farndon JR, Harris AL, Sherbet GV. Epidermal growth factor receptors
are present in human breast cancers. Br J Surg. 1984; 71: 902.
Sainsbury JRC,
Farndon JR, Harris AL, Sherbet GV. Epidermal growth factor receptors
on human breast cancers. Br J Surg 1985; 72: 186-188.
Sainsbury JRC,
Farndon JR, Sherbet GV, et al. Epidermal growth factor receptors and
oestrogen receptors in human breast cancer. Lancet 1985; l: 364-366.
Borley AC, Hiscox
S, Gee J, et al. Anti-oestrogens but not oestrogen deprivation promote
cellular invasion in intercellular adhesion-deficient breast cancer
cells. Breast Cancer Res 2008; 10: R103.
McKeage K, Perry
CM. Trastuzumab - A review of its use in the treatment of metastatic
breast cancer over expressing HER2. Drugs 2002; 62: 209-243.
Dhesy-Thind
B, Pritchard KI, Messersmith H, et al. HER2/neu in systemic therapy
for women with breast cancer: a systematic review. Breast Cancer Res
Treat 2008; 109: 209-229.
Gennari A, Sormani
MP, Pronzato P, et al. HER2 status and efficacy of adjuvant anthracyclines
in early breast cancer: a pooled analysis of randomized trials. J Natl
Cancer Inst. 2008; 100: 14-20.
Gao J, Liu YG,
Liu R, et al. Herceptin-platinum(II) binding complexes: Novel cancer-cell-specific
agents. Chem Med Chem 2008; 3: 954-962.
Phillips GDL,
Li GM, Dugger DL, et al. Targeting HER2-positive breast cancer with
Trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res 2008;
68: 9280-9290.
Cersosimo RJ.
Monoclonal antibodies in the treatment of cancer, part 1. Amer J Hlth-System
Pharm 2003; 60: 1531-1548.
Klastersky J.
Adverse effects of the humanized antibodies used as cancer therapeutics.
Current Opinion Oncol 2006; 18: 316-320.
Perez EA. Cardiac
toxicity of ErbB2-targeted therapies: What do we know? Clin Breast Cancer
2008; 8: S114-S120
McKeage K, Lyseng-Williamson
KA. Trastuzumab - A pharmacoeconomic review of its use in early breast
cancer. Pharmacogenomics 2008; 26: 699-719.
Ewer SM, Ewer
MS. Cardiotoxicity profile of trastuzumab. Drug Safety 2008; 31: 459-467.
Popat S, Smith
IE. Therapy insight: anthracyclines and trastuzumab - the optimal management
of cardiotoxic side effects. Nature Clin Pract Oncol 2008; 5: 324-335.
Bria E, Cuppone
F, Milella M, et al. Trastuzumab cardiotoxicity: biological hypotheses
and clinical open issues. Expert Opinion Biol Therapy 2008; 8: 1963-1971.
Dinh P, de Azambuja
E, Cardoso F, et al. Facts and controversies in the use of trastuzumab
in the adjuvant setting Nature Clin Pract Oncol 2008; 5: 645-654.
Nelson MH, Dolder
CR. Lapatinib: A novel dual tyrosine kinase inhibitor with activity
in solid tumors. Ann Pharmacotherapy 2006; 40: 261-269.
Johnston SRD,
Leary A. Lapatinib: A novel EGFR/HER2 tyrosine kinase inhibitor for
cancer. Drugs Today 2006; 42: 441-453.
Bilancia D,
Rosati G, Dinota A, et al. Lapatinib in breast cancer. Ann Oncol 2007;
18 Suppl 6: 26-30.
Di Leo A, Gomez
HL, Aziz Z, et al. Phase III, Double-blind, randomized study comparing
Lapatinib plus Paclitaxel with placebo plus Paclitaxel as first-line
treatment for metastatic breast cancer J Clin Oncol 2008; 26: 5544-5552.
Montemurro F,
Valabrega G, Aglietta M. Lapatinib: a dual inhibitor of EGFR and HER2
tyrosine kinase activity. Expert Opinon Biol Therapy 2007; 7: 257-268.5
Press MF, Finn
RS, Cameron D, et al. HER-2 Gene amplification, HER-2 and epidermal
growth factor receptor mRNA and protein expression, and Lapatinib Efficacy
in women with metastatic breast cancer. Clin Cancer Res 2008; 14: 7861-7870.
Sherbet GV,
Lakshmi MS. The genetics of cancer: Genes associated with cancer invasion,
metastasis and cell proliferation. 1997; Academic Press, London.
National Cancer
Institute and Eastern Cooperative Oncology Group ECOG, 2005
The National
Institute for Clinical Excellence in June 2008 guidelines.
"Pamela was assessed by the crisis team at home late at night. She had been discharged from the inpatient unit only two days previously. Pamela was threatening to set fire to herself and was wandering around her flat in her bedclothes aroused and waving a lighter. She said her bed was soaked with meths. Pamela said she had debts and there were dealers to whom she owed money who were coming round to get her. She said she did not feel safe in her flat. The crisis team were pretty sure that Pamela had been drinking and taking amphetamines and felt that she was a serious risk to herself and others. Fortunately she agreed to be admitted. By the time she arrived on the ward she was calmer and she seemed to settle quickly. A couple of days later Pamela wanted leave to go to her flat and look after her dogs. She was given 4 hours leave but did not return on time. When she returned late she seemed intoxicated. Staff felt she was abusing the ward and asked for a review with a view to discharge. When Pamela saw the doctor she said she was suicidal and was planning on taking her own life but she denied having drunk alcohol. Pamela was not willing to stay on the ward and so had to be detained she was placed on continuous observations but then managed to cut her self using a concealed razor blade while in the lavatory. Staff negotiated an agreement that Pamela would talk to them if she felt distressed and asked her to hand in any further blades. That evening Pamela had a long conversation with a junior nurse and, after some persuasion handed in some razor blades. She went back to her room and ten minutes later, again despite being on continuous observations managed to cut herself again very severely with a razor blade. The following day in the hand over Pamela was characterised as "manipulative and deceitful". The ward was very full and Pamela was "using up a bed".
Staff often feel as though they are being manipulated by patients or describe their behaviour as "manipulative" and yet this term probably adds little to the management of patients other than the expression of dislike because a wide variety of disapproved of behaviours tends to be grouped under this term. An insight into its meaning and use can be gained by noting that patients with psychosis or melancholic depression do not often attract the appellation. Instead people who are drug and alcohol dependent, people with personality disorders and some depressed patients with atypical symptoms. The key is a sense on the part of the staff that the individual has some voluntary control over their behaviour and that they could change it if they so desired. Another feature is the sense that something is being extracted or demanded in an underhand way. Another feature of situations in which patients are described as manipulative is one where resources are scarce and, for reasons of rationing staff are under pressure not to accede to requests that have resource implications.
In thinking about situations in which staff are tempted to use the idea of manipulation it is probably a good idea to distinguish three potential situations from each other.
In some situations some patients are genuinely manipulative in their intentions. That is to say they are willing to tell untruths or to create situations that persuade or force other people to do things they want without having to ask for them directly. Often patients do this when they judge that were they to ask directly they would not get what they were asking for. If Pamela had asked directly for admission to hospital to avoid her creditors and the drug dealers who were chasing her there is little doubt that this would have been refused. So some part of her aroused behaviour may have been part of a conscious plan to obtain admission to the ward where she would feel safer.
In other situations patients appear manipulative because their actions seem inconsistent or disingenuous but no covert intention exists. So, for example when Pamela says she is going home to look after her dogs that may well have been her real intent. The fact that she then got drunk and returned late is not certain to have been a conscious part of her planning when she asked to leave the ward.
A third group of situations are those in which staff feel that they have been "used" or treated in an unfair way. This is very likely to have been the case in relation to Pamela's cutting behaviour. She is sneaky,conceals blades and evades the nurse who observes her and later she reassures another nurse after receiving a good dollop of care but then immediately cuts herself again. Patients who evade ward observations make nurses understandably angry because they place the nurse at risk of censure and because they are difficult to look after. While this behaviour is annoying and while it may result in increased or prolonged periods of nursing observation it is unlikely to be manipulative. When questioned patients talk about concealing their self harm far more than they reveal it. Thus the aim of the behaviour is to evade control rather than to have an effect on staff.
A final situation in which patients can be labelled manipulative is when they are perceived to be using resources to which they have less claim than others or when they are thought not to be "taking responsibility for their behaviour". Such is the final accusation against Pamela. At such times what they do is labelled as "behavioural" or the decision made about them is that there is no evidence of genuine mental illness.
There are several serious difficulties with this way of thinking about this sort of patient. Such patients are generally thought by the public and the government to be suffering from a mental illness and characterise themselves in this way. They are clearly suffering and also clearly making a hash of their lives. Another problem with this line of thought is that it marks the end point of questioning enquiry about what is going on and often also the end point in relation to creative problem solving designed to improve the situation. Staff and the system turn from therapy to exclusion or expulsion as their main objective. Neither of these objectives turn out to be effective for psychiatric services since patients in this group return despite their ejection and, because of a failure of creative thought their problems remain in status quo.
COMPETEING INTERESTS
None Declared
AUTHOR DETAILS
CHESS DENMAN, Consultant Psychiatrist in Psychotherapy, Complex Cases Service, Springbank Ward, Cambridge And Peterborough Mental Health Foundation Trust, Fulbourn Hospital, Cambridge, CB15EF
Email: Chess.Denman@cpft.nhs.uk
Next Issue.
So what should I do then? - Managing manipulative patients.
BACKGROUND : The present study was designed to measure changes in markers of antioxidant capacity (measured individually and total) following acute ischemic stroke.
METHODS : The study included 135 subjects. 62 were controls and 73 were ischemic stroke patients diagnosed clinically and by CT scan of the brain. The cases were divided into two groups, The ischemic stroke patients with large vessel / cortical, subcortical infarcts (Group. I) and small vessel / lacunar infarcts (Group. II) based on CT scan of the brain. Serum vitamin E, vitamin C, superoxide dismutase, uric acid and total antioxidant capacity were estimated in all the subjects.
RESULTS : Group I and Group II ischemic stroke cases had significantly lower levels of vitamin E, vitamin C and superoxide dismutase and significantly higher levels of uric acid compared to controls. The group I ischemic stroke cases had significantly lower levels of vitamin E, vitamin C, and superoxide dismutase and significantly higher levels of uric acid than group II ischemic stroke cases. Total antioxidant capacity strongly correlated with serum uric acid in cases
CONCLUSION: The present study suggests that estimation of vitamin E, vitamin C, SOD, uric acid and total antioxidant capacity may be used as an indirect evidence of oxidative stress induced neuronal damage in acute ischemic stroke which may be useful for monitoring and optimizing antioxidant therapy.
KEY WORDS: Stroke, oxidative stress, SOD, vitamin C, vitamin E. Total antioxidant capacity.
Several studies provide evidence of an association between ischemic stroke and oxidative stress. Increased free radical formation together with a reduced antioxidant defense causes oxidative stress, that may play an important role in the pathogenesis of stroke associated neuronal injury. Several studies demonstrate increased oxidative damage to neuronal cells during cerebral ischemia and reperfusion. Antioxidant activity is known to reflect the altered redox balance of affected fluids, tissues or organs in acute ischemic stroke patients. Therefore antioxidant concentrations or measures of their activity have been used to estimate the amount of oxidative stress 1. No single component of serum antioxidant complex could fully reflect the protective efficiency of blood, probably because of interactions that occur in vivo among different antioxidant compounds. Total antioxidant capacity considers the cumulative effect of all antioxidants present in blood and body fluids 2. The aim of this study was therefore to measure changes in markers of antioxidant capacity (measured individually and total) following acute ischemic stroke.
Materials and methods:
This study was conducted at Bapuji Hospital and Chigateri General Hospital, Davangere (Both Hospitals attached to J.J.M. Medical College, Davangere, karnataka), by including 62 healthy controls (of which 34 were men and 28 were women aged between 36 and 73 years) and 73 cases clinically diagnosed
as acute ischemic stroke patients of less than 48 hrs duration after the onset of symptoms) and confirmed by computerized tomography of the brain (of which 41 were men and 32 were women aged between 36 and 73 years). The cases were divided into two groups, The ischemic stroke patients with large vessel / cortical, subcortical infarcts (Group. I) and small vessel/lacunar infarcts (Group. II) based on CT scan of the brain. The stroke patients due to cerebral hemorrhage, malignancy, sepsis, severe medical or psychiatric illness, language disorders, swallowing difficulties, cognitive impairment, gout, renal failure and patients who were taking antioxidant vitamins were excluded from the study. The study was conducted after informed consent was obtained from them and the study has been approved by the ethical committee of the institution.
Under aseptic precautions about 6 ml of a non-fasting venous blood sample was collected from cases within 24 h following stroke onset and from healthy controls. Blood was collected in appropriate tubes and centrifuged at 3000 g for 15 min to separate plasma from red blood cells. The supernatant was stored at 40 C until analysis was carried out.
Serum vitamin E was estimated by Baker and Frank method 3, Vitamin C by 2, 4 DNPH method 4, SOD by Marklund and Marklund method 5, and uric acid by Henry Caraway method 6 and total antioxidant capacity (TAC) by FRAP assay method 2 in both the controls and cases. All the chemicals used were of highest analytical grade available in India.
TABLE I. Comparison of Serum Vitamin E, Vitamin C, SOD, Uric Acid and Total Antioxidant Capacity (Mean SD)
Vit. E mg/L
Vit. C mg/dl
SOD units/ml
Uric acid mg/dl
TAC (mol/l)
Controls (n = 62)
11.04 0.97
1.16 0.13
9.01 1.03
4.66 0.47
1079.7 197.9
Cases (n = 73)
7.22 0.81
0.52 0.16
4.35 0.70
6.56 0.73
1043.4 140.7
Comparison
p < 0.05
p < 0.05
p < 0.05
p < 0.05
p > 0.05
TABLE II. Comparison of Serum Vitamin E, Vitamin C, SOD, Uric Acid and Total Antioxidant Capacity (Mean SD) between Group I and Group II Cases.
Vit. E mg/L
Vit. C mg/dl
SOD units/ml
Uric acid mg/dl
TAC (mol/l)
Cases Group. I (n = 56)
6.93 0.67
0.45 0.08
4.14 0.61
6.78 0.62
1048.9 140.3
Cases Group. II (n = 15)
8.24 0.17
0.80 0.10
5.12 0.49
5.73 0.55
1038.1 142.9
Comparison (one way ANOVA and student t test)
p < 0.05
p < 0.05
p < 0.05
p < 0.05
p > 0.05
Statistical analysis :
Statistical analysis was performed with one way ANOVA test, student t test and Pearsons correlation coefficient using SPSS version 16.0. A value of p < 0.05 was taken to indicate statistical significance.
Results:
It was observed that the serum levels of Vitamin E, Vitamin C, TAC and SOD were significantly lower in ischemic stroke cases than those of controls and serum uric acid levels were significantly higher in ischemic stroke cases table I . Further it was observed that the group I ischemic stroke cases had significantly lower levels of serum Vitamin E, Vitamin C and SOD than group II ischemic stroke cases and significantly higher serum levels of uric acid in group I cases than group II ischemic stroke cases table II . Significant negative correlations were observed between vitamin C, vitamin E, SOD and TAC and significant positive correlation was observed between uric acid and TAC among cases table III.
TABLE III. Correlations of Vitamin E, Vitamin C, SOD, Uric Acid with Total Antioxidant Capacity in cases.
Vitamin C and TAC
Vitamin E and TAC
SOD and TAC
Uric acid and TAC
R value
-0.24
-0.1
-0.19
0.16
p value
< 0.05
< 0.05
< 0.05
< 0.05
Discussion:
In this study, there were reduced concentrations of vitamin E, vitamin C, SOD and TAC and increased concentrations of uric acid in stroke patients compared with controls. FRAP assay is presented as a novel method of assessing total antioxidant capacity 2 which is believed to be a useful measure of the ability of antioxidant present in the fluids to protect against oxidative damage to membranes and other cellular components..
Vitamin E, a potent chain breaking lipid soluble antioxidant, reacts with lipid peroxyl radicals eventually terminating the peroxidation chain reaction and thereby reducing oxidative damage. Some studies have shown reduced serum vitamin E levels in stroke patients and this may be due to high lesion volume resulting in production of more number of free radicals from a large ischemic injury. It is also shown that reduced vitamin E levels resulted in poor clinical outcome in stroke patients 7,8. In the present study serum vitamin E levels were significantly decreased in ischemic stroke cases (significantly decreased in large vessel infarcts than in small vessel infarcts) when compared to controls.
Vitamin C represents the major water-soluble antioxidant in the human body. Many studies show that reduced vitamin C levels are associated with increased risk of both ischemic and hemorrhagic strokes 9. In our present study the serum vitamin C levels were decreased significantly in ischemic stroke cases (decreased significantly in large vessel infarcts than in small vessel infarcts) compared to controls. It may be due to the exhaustion of this antioxidant in the neutralization of free radicals which are formed in excess during ischemia and reperfusion 10,11.
SOD is an endogenous antioxidant that catalyses the dismutation of the superoxide anion radical. SOD plays an important role in the defense against free radical damage in reperfusion injury and helps in reducing the infarct size during ischemia and reperfusion 12,13. In the present study the serum SOD levels were decreased significantly in ischemic stroke cases decreased in large vessel infarcts than in small vessel infarcts) compared to controls.
Uric acid, most abundant endogenous aqueous antioxidant in humans, may protect against oxidative modification of endothelial enzymes and preserves the ability of endothelium to mediate vascular dilatation during oxidative stress 14. Several studies have shown that increased oxidative stress is associated with high circulating uric acid levels due to elevation of xanthine oxidase in stroke induced brain damage 15,16. In this study, there was a significant increase in the serum levels of uric acid in ischemic stroke cases (increased significantly in large vessel infarcts than in small vessel infarcts) than in controls. Serum TAC strongly correlated with serum uric acid. Under multivariate analysis, serum uric acid explained most of the variance in TAC during the study period.
This study suggests that estimation of serum vitamin E, vitamin C, SOD, uric acid and total antioxidant capacity may be used as an indirect evidence of oxidative stress induced neuronal damage in ischemic stroke.
The limitations of our study are as follows: We have not estimated any markers of lipid peroxidation such as malondialdehyde which along with antioxidant levels would better explain oxidative stress. Antioxidant levels were measured only once, but prospective serial estimations would better predict the antioxidant status with prognosis of stroke. This study was conducted on a small group of stroke patients. Larger clinical studies in this area are needed to establish the relationships between antioxidant capacity and oxidative damage following ischemia and reperfusion in man, and to form the basis of appropriate antioxidant intervention strategies to minimize long-term brain injury following cerebral ischemia.
COMPETING INTERESTS
None Declared
AUTHOR DETAILS
DR. SRIKRISHNA R, M.D., Assistant Professor, Department of Biochemistry, JJM Medical college, Davangere, India
DR. SURESH D R, M.D., Assistant Professor, Sri Siddhartha Medical College, Sri Siddhartha Academy of Higher Education (SSAHE), Tumkur, India
CORRESPONDENCE: DR. SURESH D R, 3/1, Seethappa layout, 5th Block, Doddabommasandra, Vidyaranyapura.P.O, Bangalore, Karnataka, INDIA- 560097.
Email: drsuri77@gmail.com
References
Leinonen JS, Ahonen JP, Lonnrot K, Jehkonen M, Dastidar P,Molnar G. Low plasma antioxidant activity is associated with high lesion volume and neurological impairment in stroke (2000). Stroke 31(9), 33-39.
Benzie FF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of antioxidant power: the FRAP assay (1996). Analytical Biochem 239, 7076.
Baker H, Frank O (1969). Clinical vitaminology. Academic Press. New York, pp169173.
Omage ST (1979). Ascorbic acid analysis II. Determination after derivatization with 2, 4, - Dinitrophenylhydrazine, Methods in Enzymology, 62, 7-8.
Nandi A, Chatterjee IB (1988). Assay of superoxide dismutase activity in animal tissues. J Biosci, 13 (3), 305-315.
Price CP, James DR (1988). Analytical reviews in clinical biochemistry: the measurement of urate. Ann Clin Biochem, 25 (5), 484-498.
Cherubini A, Ruggiero C, Polidori MC, Mecocci P. Potential markers of oxidative stress in stroke. Free Radical Biology and Medicine (2005), 39 (7), 841-852.
Tornwall ME, Virtamo J, Korhonen PA, Virtanen MJ, Albanes D, Huttunen JK (2004). Postintervention effect of alpha tocopherol and beta carotene on different strokes. A 6 year follow-up of the alpha tocopherol, beta carotene cancer prevention study. Stroke, 35(8), 1908-1913.
Yokoyama T, Date C, Kokubo Y, Yoshiike N, Matsumura Y, Tanaka H (2000). Serum vitamin C concentration was inversely associated with subsequent 20 year incidence of stroke in a Japanese Rural Community. Stroke, 31(10), 2287-2294.
Kurl S, Tuomainen TP, Laukkanen JA, Nyyssonen K, Lakka T, Sivenius J, Salonen JT (2002). Plasma vitamin C modifies the association between hypertension and risk of stroke. Stroke, 33(6), 1568-1573.
Kim GW, Kondo T, Noshita N, Chan PH (2002). Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia / reperfusion in mice. Stroke, 33(3), 809-815.
Zimmermann C, Winnefeld K, Streck S, Roskos M, Haberl RL (2004). Antioxidant Status in Acute Stroke Patients and Patients at Stroke Risk. Eur Neurol, 51(3), 157-161.
Weir CJ, Muir SW, Walters MR, Lees KR (2003). Serum urate as an independent predictor of poor outcome and future vascular events after acute stroke. Stroke, 34 (8), 1951-1957.
Hariklia VD, Apostolos IA, Haralambos IK (2008). The Role of Uric Acid in Stroke: The Issue Remains Unresolved. Neurologist, 14(4), 238-242.
Hozawa A, Folsom AR, Ibrahim H, Nieto JF, Rosamond WD, Shahar E (2006). Serum uric acid and risk of ischemic stroke : the ARIC study. Atherosclerosis, 187(2), 401-407.
Bullying in the workplace is emerging as a problem over the past decade. Despite the tendency for incidents of bullying to be underreported 2 it is widespread in all sectors of the workforce including healthcare in the United Kingdom (UK) 3. The culture of bullying in medicine contributes to this pattern of bullying behaviour that can adversely affect any aspect of working life from an employees health 4 to the reputation of the organisation 5. Therefore immediate changes are required to increase the recognition of this problem and take further steps to a solution.
Bullying and harassment
There are different ways to understand the terms bullying and harassment but considerable overlap exists with similar patterns of behaviour (figure 1).
Figure 1: Examples of bullying and harassment 6
Spreading malicious rumours, or insulting someone by word or behaviour (particularly on the grounds of age, race, sex, disability, sexual orientation and religion or belief)
Copying memos that are critical about someone to others who do not need to know
Ridiculing or demeaning someone picking on them or setting them up to fail
Exclusion or victimization
Unfair treatment
Overbearing supervision or other misuse of power or position
Unwelcome sexual advances touching, standing too close, the display of offensive materials, asking for sexual favours, making decisions on the basis of sexual advances being accepted or rejected
Making threats or comments about job security without foundation
Deliberately undermining a competent worker by overloading and constant criticism
Preventing individuals progressing by intentionally blocking promotion or training opportunities.
The essential difference between bullying and harassment is that the latter is usually a single incident that relates to ones social identity and is therefore viewed as discriminatory in nature e.g. racial or sexual harassment. In legal terms harassment refers to a course of conduct directed at a specific person, which causes substantial emotional distress, and can be identified by equality laws in the relevant country.
On the other hand workplace bullying is generally not covered by specific legislation. The exception to this is found in such as Sweden and Norway 7. Indeed it is in Scandinavia where extensive research into bullying in the workplace originated 7.
Bullying
Bullying in the workplace is known internationally by terms such as mobbing, workplace harassment, employee abuse, mistreatment at work, and petty tyranny 8. There is no generally accepted definition of workplace bullying but it is summed up well by the following:
Persistent, offensive, abusive, intimidating or insulting behaviour, abuse of power or unfair penal sanctions which makes the recipient feel upset, threatened, humiliated or vulnerable, which undermines their self-confidence and which may cause them to suffer stress 9.
It is important to distinguish between bullying, which is always undermining and destructive, and constructive supervision that is developmental and supportive 8. The three essential elements of bullying are that it has a negative impact on the victim, it is persistent and, crucially, bullying is subjective 10. If a person feels bullied then he/she is being bullied 11. This last point may be controversial because it is dependent on the bullied persons views and not based on objective evidence. Nevertheless workplace bullying exists as a problem. According to the Chartered Institute of Personnel and Development (CIPD) there has been a shift of perception in organisations from denying it happens to accepting that bullying is a problem 3.
How common is bullying?
The Silent Epidemic 7
Workplace bullying affects up to 50 per cent of the UK workforce at some time in their working lives and has an annual prevalence nearly 40 per cent 7. One in 10 callers to the UK National Bullying Advice Helpline are health care professionals 3. A questionnaire survey 12 revealed that 38% of staff in a community healthcare trust were subject to workplace bullying in the previous year and that 42% had witnessed bullying of others. The British Medical Association (BMA) has acknowledged that bullying rates are higher in healthcare organisations and stated that 1 in 7 National Health Service (NHS) staff reported being bullied by other staff 13.
The scale of the problem has been widely highlighted as a problem in the nursing profession 10 with increased rates of bullying reported in Black and Minority Ethnic (BME) groups 14. In doctors bullying may occur in the clinical, educational 8 and research environment 15. One survey of doctors in the UK revealed that 37% of junior doctors had been bullied and 84% had experienced at least one bullying behaviour in the preceding year16. Higher rates have been reported in non-European Union (non-EU) doctors practicing in westernised countries 17 who are also less likely to take action against bullying 18.
Despite the growth of literature in this area the problem of workplace bullying is obscured by underreporting which has numerous causes (figure 2).
Figure 2: Reasons for underreporting of bullying 2
Fear it will make matters worse
The belief that nothing would be done about it
Concerns about confidentiality
Fear of possible victimisation
Concerns of being labelled a troublemaker
May be seen as an admission of failure
A degree of learned tolerance that may imply that the behaviour is acceptable
The greatest fear is that of reprisals from the employer, associates of the bully, and powerful professionals, who may close ranks and compromise the career of the whistle blower 1.
Why do people bully in medicine?
The antecedents to bullying have undergone considerable debate in the psychology literature. Bullies may be attracted to the caring professions to take advantage of the vulnerability embedded in them in relation to clients and employees 1. However in most cases the bullying in medicine is likely to be unintentional and could be shaped by the power inequality in relationships (e.g. consultant Vs junior doctor) in the field.
Moreover the traditional hierarchy within medicine and the teaching by intimidation and humiliation may foster a culture of bullying 18. Studies in the United States 19 and UK 13 have suggested that bullying commences with medical student and that this sets up a transgenerational legacy 7 as the behaviours of bullying are passed down. The BMA urges for a stop to the cycle of bullying and argue further that the target ethos in the health service with the survival of the fittest culture adds to bullying 13.
How do you know if you are being bullied?
If you are being bullied early warning signs may be present. These include the perception that your working relationship is different, that you are being persistently got at, that your work is being unfairly criticised, or you begin to question whether these mistakes you are supposed to have made really are your fault 20. In addition to feelings of being undermined, or humiliated, bullying may also be associated with symptoms (figure 3).
Figure 3: Symptoms of bullying 20
Physical
Emotional
Sleeplessness
Acute anxiety
Nausea
Feeling isolated
Migraine/severe headaches
Loss of confidence/self-esteem
Palpitations
Depression
Skin complaints
Panic attacks
Sweating/shaking
Anger
Stomach problems
Mood swings
Backache
Lack of motivation
Loss of appetite
Suicidal thoughts
Lethargy
Why does bullying matter?
It is clear from the physical and psychological effects that bullying affects people in their personal health. Workplace bullying can also contribute to problems of staff retention and economy. Estimates suggest that in the UK bullying cost employers 80milion lost working days and up to 2-30 billion in lost revenue each year 7. It costs the NHS more than 325 million a year and accounts for around 50 per cent of stress-related workplace illnesses 5.
Other effects of bullying at work include poor morale, poor employee relations, loss of respect for managers or supervisors, poor performance, lost productivity, absences, resignations, damage to organisations reputation and potential costs in tribunal and other court cases 6. Ultimately if the culture of bullying results in demoralized staff working, in a caring profession, it is the patients who will suffer.
What is currently being done about it?
In the UK the BMA has called for zero tolerance on bullying 13 and have provided a report on bullying and harassment in the workplace 21. Most NHS trusts disseminate anti-bullying policies, in connection with Dignity at Work, but the effective implementation of these policies has been questioned with the criticism that it is only for show 18. The information on guidance and policy, in relation to workplace bullying, is not widely publicised and the question is whether bullying is being systematically played down?
Recommendations
Although organisations such as the health service have taken steps to deal with bullying it is clear that problems persist. Heenan 5 states that an all-singing all-dancing policy is worthless without a culture that believes in and supports it and recommends steps employers need to consider (figure 4).
Figure 4: Key steps recommended for employers 5
Look at the culture of the organisation where and how might the risk of harassment arise?
Foster an environment where staff feel able readily to raise any concerns, before they become problems.
To support this, have a clear and well publicised policy to tackle harassment issues.
Back this up with training (including how to handle grievances) and set good examples through role models.
Deal with harassment wherever and however it arises, to demonstrate that it is unacceptable and will not be tolerated.
Provide independent employee assistance, including confidential counselling and other support for employees to enable to challenge unreasonable behaviour which, left unchecked, could lead to harassment.
Figure 5: What to do if you are being bullied 2
Steps to take
Options for support
Approach, or write to, the bully and ask them to stop
Speak to a friend, colleague, supervisor or manager
Ask line manager, supervisor, human resource representative or trade union official to speak to the bully.
Ask employer for support from a specially trained staff member
Keep a record of any incidents and informal action taken
Speak to general practitioner especially if your health is affected
Consider a formal complaint in writing to their line manager or human resources representative
Seek counselling which has been provided by the NHS to its entire staff since 2000
Have a colleague accompany you to any formal investigation meetings
Contact bullying and harassment hotlines
Formal investigation may recommend a disciplinary hearing
Employer may refer you to an external agency for more support
Alternative management action may be considered e.g. facilitated discussion or redeployment
Mediation may be on offer to encourage and help reach an informal outcome
Awareness of bullying needs to be raised and the problem dealt with at an organisational and individual level. The authors suggest that bullying should be incorporated into teaching programmes and induction of junior doctors. Heenan 5 recommends training for managers and supervisors so that they have the confidence to deal with a situation, and deal with it at an early stage, rather then allowing the problem to accumulate and end up in the courts. Therefore it is in the healthcare trusts interests to take these steps to monitor and manage this problem.
In addition employees in healthcare need to be better informed of what steps to take if they find themselves as victims of a bully at work. NHS employers provide options available to deal with bullying and provide support for it (figure 5).
Conclusion
The late Tim Field, founder of the National Workplace Bullying Helpline, warns that everyone is at risk of becoming a target of bullying 1. However the bully in healthcare organisations may not often realise what they are doing, so do both parties require help? There are conflicting views for the solution to bullying in the workplace regarding whether educational 22 or punitive 17 measures are appropriate. This will continue to be a matter of debate. Whichever approach is adopted, identification and increased awareness of bullying is the first step to the solution.
Bullying is an old problem that keeps re-emerging without a clear solution 3
KEY POINTS
Bullying is subjective if you feel bullied then you are bullied
Bullying is more prevalent then we think because of underreporting
Causes of bullying are complex and may be embedded in the culture of the organization
Being bullied is associated with emotional and physical symptoms
Bullying has implications at a personal, social, and organisational level
Implementation of policies by health care trusts need to be improved
Organisations need to be more proactive in raising awareness of this growing menace and demonstrate that it is unacceptable
JAVED LATOO, MBBS, DPM, MRCPsych, North East London NHS Foundation Trust, United Kingdom
CORRESPONDENCE: Dr MINAL MISTRY, Hampshire Partnership NHS Trust, Melbury Lodge, Winchester, United Kingdom
Email: minalmistry@yahoo.co.uk
References
1. Field T, Becker K, Mackenzie GM, and Crossan L. Bullying in medicine. BMJ. 2002; 324: 786.
2. NHS Employers. Bullying and harassment staff guidance. 2007. Available from: http://www.nhsemployers.org/HealthyWorkPlaces/BullyingAndHarassment/Pages/Staffguidance.aspx
3. Al-Daraji WI. An old problem that keeps re-emerging without a clear solution. Medico-Legal Update. 2008; 8(2), 24-30.
4. Kivimki M, Elovaino M and Vahtera J. Workplace bullying and sickness absence in hospital staff. Occup Environ Med. 2000; 57: 656-660.
5. Heenan R. How to beat the workplace bully. Health Service Journal. 12th February 2009: 25-27.
6. ACAS advice leaflet. Bullying and harassment at work: guidance for employees. 2008.
7. McAvoy B and Murtagh J. Workplace bullying the silent epidemic. BMJ. 2003; 326: 776-777.
8. Hicks B. Time to stop bullying and intimidation. Hosp Med. 2000; 61 (6): 428-431.
9. Lyons R, Tivey H, and Ball C. Bullying at work: how to tackle it. A guide for MSF representatives and members. London: MSF. 1995.
10. Quine L. Workplace bullying in nurses. J Health Psych. 2001; 6: 73-84.
11. Macpherson W. Stephen Lawrence inquiry: report of an enquiry by Sir William Macpherson of Cluny. London: Stationary Office. 1999.
12. Quine L. Workplace bullying in NHS community trust: staff questionnaire survey. BMJ.1999; 318: 228-232.
13. BMA Newswire article. BMA calls for zero tolerance on bullying and harassment in the Workplace. 19 May 2006.
14. Giga S, Hoel H, and Lewis D. A review of Black and Minority Ethnic (BME) employee experiences of workplace bullying. University of Bradford, (Research commissioned by the Dignity at Work Partnership). May 2008.
15. Stebbing J, Mandalia S, Portsmouth S, Leonard P, Crane J, Bower M, Earl H and Quine L. A questionnaire survey of stress and bullying in doctors undertaking research. Postgrad Med J. 2004; 80: 93-96.
16. Quine L. Workplace bullying in junior doctors: questionnaire survey. BMJ. 2002; 324: 878-879.
17. Cheema S, Ahmad K, Giri SK, Kaliaperumal VK, Naqvi SA. Bullying of junior doctors prevails in Irish health system: a bitter reality. Ir Med J. 2009; 98(9):274-5.
18. Hoosen A and Callaghan R. A survey of workplace bullying of psychiatric trainees. Psych Bull. 2004. 28: 225-227.
19. Frank E, Carrera JS, Stratton T, Bickel J, and Nora LM. Experiences of belittlement and harassment and their correlates among medical students in the United States: longitudinal survey. BMJ. 2006; 333:682
20. Andrea Adams Trust. Factsheet on workplace bullying. 1997. Available online from: http://www.andreaadamstrust.org/live/factsheet.html
21. BMA report. Bullying and harassment of doctors at work in the workplace, 17 May 2006. Available online from: http://www.bma.org.uk/employmentandcontracts/morale_motivation/bullying2006.jsp
22. Paice E, Aitken M, Houghton A, and Firth-Cozens J. Bullying among doctors in training: cross sectional questionnaire survey. BMJ. 2004. 329: 658-659.
Society of Hospital Medicine (SHM) defines 'Hospitalists' as physicians whose primary professional focus is the general medical care of hospitalized patients. Their activities include patient care, teaching, research, and leadership related to hospital medicine.
The term "hospitalist"1was first introduced in 1996 by Robert M. Wachter and L Goldman to describe physicians who devote much of their professional time and focus to the care of hospitalized patients. In the most prevalent American model of hospitalist care, several doctors practice together as a group and work full-time caring for inpatients. Most of the (80%) of practicing hospitalists are board certified or eligible in internal medicine, and some (5%) have completed subspecialty fellowships. Although hospitalists first emerged in the care of adult inpatients, the field has grown rapidly in pediatrics, now accounting for nearly 10% of U.S. hospitalists2. Hospitalists typically provide 24/7 inpatient coverage and thus are more readily available to a patient than a doctor who spends much of the day outside the hospital in an office or clinic setting.
Over the past decade, the United States has undergone a remarkable evolution in the way it delivers inpatient medical care. In the mid-1990s, much of American health care was dominated by a managed care paradigm, which gave incentives to control health care inflation3. Hospitalist model was uniquely well versed in evidence based practice and systems improvement. Their focus on providing clinically appropriate care, improving efficiency, reduce length of inpatient stay and helping to make the hospital system work better, without compromising patient satisfaction & outcome was big boon for its growth. Hospitalist field has now become the fastest growing specialty in the history of American medicine, approximately today close to 15,000 hospitalists practice in America, and the field is likely to grow to about 30,000 making it a larger specialty than cardiology4.
Hospitalist Program, St Joseph's Hospital/Marshfield Clinic
In keeping pace with nation wide trend, St. Joseph's Hospital (SJH) Marshfield Clinic, Marshfield was developed in 2000 by the General Internal Medicine Department at the request of clinic leadership. Hospitalst Program was formally launched in October 2001. SJH was pioneer and leader during the time in the Midwest. Dr Qasim Raza & Dr Mark Schwartz were instrumental in establishing a full fledged hospitalist program that has grown tremendously over the years. Dr Bill Yanke, then Chairman of Internal Medicine made into a full subdivision in October 2005 under the leadership of Dr Qasim Raza as Medical Director and Dr Mark Schwartz as Associate Program Director. Today there are 24 full-time hospitalists providing 24/7 hours inpatient medical services, including consultative services to all sub-specialists at Marshfield Center. Their job also includes surgical co-management of all orthopedic patients admitted to St. Josephs hospital.
Hospitalists typically work in shift system, and on any given day we have about 7 day time non-teaching, 2-3 academic teaching, 1 back-up, 2 evening and 1 night time hospitalist covering all medical inpatient services. St. Joseph's Hospital/Marshfield has always been able to recruit the top graduating residents from Internal Medicine Residency/Med-Peds Programs across United States as we believe this is the first step towards success of our program. Many of our hospitalists are actively involved in academic research & teaching and hold Clinical Assistant Professor rank with School of Medicine & Public Health, University of Wisconsin, Madison.
Andy Weir, Director Quality & Strategic Analysis, St. Joseph's hospital (SJH), Ministry Health places our program among the top 15 in the State of Wisconsin. SJH continues to have a lower Length of Stay (LOS) for the top 25 DRG (Diagnosis Related Group) that usually makes typical hospitalist patients. In FY 2007 we still held a 0.16 LOS advantage compared to other 14 hospitals that round up the top 15 hospitals of Wisconsin. This literally means thousands of dollars saved for the patients in todays world of skyrocketing health care costs. This was achieved without significant compromise on quality of care or patient satisfaction. We have far few readmits when compared to our peers in other hospitals of the state. This was only achieved with hard work and dedication of our hospitalist team under the able leadership of Dr Roderick Koehler, Chairman of Internal Medicine Department and full support of Marshfield Clinic Board of Directors.
Marshfield Clinic physicians work in four other hospitalist programs besides St Joseph's Hospital/Marshfield Center. Marshfield Clinic Euclaire, Minocqua & Wausau Centers have 4 full time hospitalists each. Marshfield Clinic Lake View Medical Center; Rice Lake has one full time hospitalist. This makes Marshfield Clinic almost the largest employer of Hospitalists (total of 37 today) in Wisconsin.
The Future : Hospitalists are here to stay
Proponents say Hospitalists fill a growing gap in continuity of patient care. Typically Physicians spend more time today in treating patients in their offices than at hospitals. Hospitals are traditionally getting sicker patients than ever before, and no primary are physicians are willing to take care of unassigned patients (patients with no primary care provider privileged to work in their hospital). Hospitalists are easily available 24 hours daily to take are of these acutely ill patients. Primary care physicians and sub-specialists are happy in that they can spend more time in their practices. There is no competition as Hospitalists have no out-patient practice and their patients return back for follow-up appointments. Its a win-win situation for patients and physicians. Medicare and more insurance companies have now tagged reimbursement with quality of care provided to patients. Research has proven that in-house physicians are good for hospitals goal to achieve these targets5.
Table 1 Potential roles for hospitalists. ( Swiss Med Wkly 2006; 136:591-596 )
Clinical
Inpatient Wards
Intensive Care Unit
Medicine Consultation Services
Palliative Care Services
Post-discharge Clinic Services
Pre-operative Clinic Services
Non-teaching services (in Teaching Hospitals)
Skilled Nursing Facilities
Educational
Residency Program Directorship
Student Clerkship Directorship
Curriculum Development and Leadership
Operational
Emergency Department Triage Officers
Bed Flow Coordination
Discharge Planning Coordination
Transfer Center Coordination
Quality & Safety
Patient Safety Officer
Director of Quality (Compliance)
Quality Improvement Officer
Other
Clinical Information Technology Implementation
Hospital Leadership Positions
Dr Robert Watcher in his landmark article in JAMA concluded that implementation of Hospitalist programs was associated significant reductions in resource use, usually measured as hospital costs (average decrease , 13.4%) or average LOS (average, 16.6%). All research till date has empirically proven that hospitalists improve in-patient efficiency without harmful effects on quality or patient satisfaction6.
Hospitalists come from diverse training backgrounds and hence SHM has started implementing a process to start early training programs for hospitalists, including residency track and fellowship programs. Current educational deficits include training in communication skills, end-of-life care, quality improvement and patient safety, medical economics, follow-up of acute post-op surgical patients. Hospitalists are slowly expanding into other roles beyond the traditional role of medical consultant Table 1.
Hospital Medicine : New Specialty
Specialists in medicine are traditionally defined by organ disease, example Cardiology; Gastroenterology, Nephrology, Radiology, Oncology, General Surgery etc. The hospitalist, on the other hand is a site defined generalist specialist similar to ER physicians. They care for acutely ill patients with wide array of organ derangements and ages in a given specific location7. Hospitalist co-ordinate and integrate patient care within the health delivery system and reduce the distance between office and hospital with their round of clock availability. Hospitalists already have their own Clinical Textbook8 and SHM is the fastest growing medical society in United States and soon plan to get credentials from ABME (American Board of Medical Specialties) to start an accredited fellowship program in Hospital Medicine.
Critical Issues Facing Hospitalists today
There is significant variation in hospitalists training level, and in the way hospitalists groups are managed. Starting a Hospitalist core curriculum in Residency Training program is the key step towards this goal. Marshfield Clinic Internal Medicine Residents already do Hospitalist Service elective rotation for one month during their post-graduate training period at SJH. Funding of hospitalist programs remains a challenge for hospital administrators. Stagnant Medicare reimbursement, rise in uninsured patients, more acutely ill patients are increasing costs, while their ability to support these costs from hospital budgets is decreasing. There is increasing diversity of hospitalists clinical and non-clinical duties, burn out myth due to ever increasing work-loads, rise in demand for hospitalists (1 available for 10 jobs!), perceived shortage may potentially compromising the efficiency and advantages of hospitalists9. Division between out-patient and inpatient practices continues to widen, hence it is vital to maintain connections between referring and primary care physicians. All of these may in-turn negatively impact hospiltalist and patient satisfaction10.
Respondents (patients and allied health care providers) have overwhelming positive impressions of hospitalist movement. In one survey over 76% believed that they improve Emergency Room efficiency, and 66% felt hospitalists lower costs. Interestingly majority 69% would prefer hospitalists have additional certification or training. In 2007 at least 59% and probably closer to 2/3 of California Hospitals have hospitalists. I believe its true for Wisconsin as well. Quality improvement, keeping patients satisfaction (health care customer), challenging all the above critical issues discussed will be the key to success of this hospitalist program at St. Josephs Hospital/Marshfield Clinic in future.
The impact of this revolution in Inpatient care is just beginning to be felt, and history will tell us if this is the best thing that has happened to medicine this decade. Nevertheless, hospital care will likely remain a highly pluralistic system in which organization of care is determined by efforts to improve the value of care in context to local demands and needs11. I am sure everybody will agree that, method of care chosen should always be the one that promotes the best clinical outcomes and highest patient satisfaction at lowest costs. With these goals hospitalists will definitely have increasingly visible role in many institutions across the country in near future.
COMPETING INTERESTS
None Declared
AUTHOR DETAILS
MOHAMMED MOIZUDDIN MD, Clinical Assistant Professor of Medicine, School of Medicine & Public Health, University of Wisconsin, Madison. Hospitalist, St. Josephs Hospital/Marshfield Clinic, Marshfield, USA
QASIM RAJA, MD, Director, Hospitalist Division, Department of Internal Medicine, Marshfield Clinic, Marshfield, USA
QURETUL QURESH, MD, Deccan College of Medical Sciences, India
CORRESPONDENCE: MOHAMMED MOIZUDDIN, MD, Department of Internal Medicine, Hospitalist office 3J3, Marshfield Clinic, 1000 North Oak Avenue, Marshfield, USA, WI 54449
Email: moizuddin.mohammed@marshfieldclinic.org
References
1. Watcher RM, Goldman L. The emerging role of "hospitalists" in the American health care system. N Engl J Med. 1996; 335:514-7.
2. Bellet PS and Watcher RM. The hospitalist movement and its implications for care of hospitalized children. Pediatrics 1999; 103:47377
3. Committee on Quality of Health Care in America, Institute of Medicine. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington DC: National Academy Press, 2001.
4. Niraj L Sehgal, RM Watcher The emerging role of "hospitalists" in the American health care system. Swiss Med Wkly 2006; 136:591-596
5. Hospitals & Health Network; June 2006, Vol. 80 Issue 6, p56-60, 3p
Where several different objects produce the same effect, it must be by means of some quality, which we discover to be common amongst them. For as like effects imply like causes, we must always ascribe the causation to the circumstance, wherein we discover the resemblance.
David Hume, A Treatise of Human Nature
The present findings suggest that even if everyone was treatedin the best possible fashion, about 60% of the burden of mentaldisorders appears to be unavertable in the light of currentknowledgeeven withperfectcoverage and treatment, half the burden of anxietydisorderswould remain unavertable.
Andrews G, Issakidis C, Sanderson K, Corry J, Lapsley H: Utilising survey data to inform public policy: comparison of the cost-effectiveness of treatment of ten mental disorders. Br J Psychiatry 2004; 184:526533
Psychiatric conditions tend to cluster together so that if a person suffers from one neurotic psychiatric disorder that person is significantly more likely to simultaneously suffer from another, so called comorbidity, and sufferers from psychiatric disorders are more likely to suffer psychiatrically again in the future in comparison to a random population.
The situation in respect of post traumatic stress disorder (PTSD) might be expected to be somewhat different because, atypically, in this disorder we presume that we know the aetiology and it is a necessary diagnostic criterion that a person has been exposed to an unusually threatening event. Nevertheless, there is recent evidence that the likelihood of recurrence of PTSD is high, despite the low frequency of catastrophic precipitating events. The various symptomatic manifestations, upon which specific psychiatric diagnoses are based, may be surface phenomena of a unifying underlying predisposition with common biological substrates. This would accord with the fact that antidepressants are effective not only in psychotic depression, melancholic type depression and non-melancholic depression but confer a wide spectrum of additional therapeutic benefits, including benefit in panic disorder, phobic disorders, obsessive compulsive disorder and PTSD.
Alternative or aggravating factors are that people who suffer from a neurotic disorder or disorders tend both to complain more and to attract adversities. These issues will be discussed in relation to the psychological concepts of neuroticism and recent biological factors.
Recurrence of disorders
Depression
Once initiated depression tends to recur (DSM - IV), in his review, Judd1 found that about 80% of persons experiencing a major depressive episode will have at least 1 more such episode during their lifetime, with the rate of recurrence being even higher if minor episodes are included. The review by Judd1 is close to the more recent estimate of Andrews2 that over the 10 years following a depressive episode, 75% of patients experience a recurrence2. Depression resulting as a reaction to stress (in which environmental demands are perceived as exceeding resources) is particularly likely to pursue a chronic and relapsing course3;4.
Anxiety Disorders
Anxiety is often an appropriate emotion which should lead to behaviour which reduces the anxiety rather than a disordered state of frozen inactivity or maladaptive actions which exacerbate the anxiety. Anxiety disorders are known to be familial and heritable5 and tend to be persistent. They can be associated with depression or be the prelude to depression6. The severity of symptoms may vary with time and be less problematic in calm, uneventful circumstances but panic disorder with or without agoraphobia often remains a chronic condition7;8.
PTSD
The definition of PTSD stands in contrast to adjustment disorder. In DSM IV an adjustment disorder is characterised by a disproportionate reaction to the stressor and therefore probably indicative of prior psychiatric vulnerability. This interpretation is reinforced in ICD 10; Individual predisposition or vulnerability plays a greater role in the risk of occurrence and the shaping of the manifestations of adjustment disorders than it does in the other conditions in F43 (Acute Stress Reaction). However, the notion that certain experiences can trigger a disproportionate reaction in some people by virtue of some perhaps occult vulnerability exhibiting a much greater susceptibility, a disproportionate effect, was commented on in the second World War for conditions which we would now diagnose as PTSD (e.g.9). An early Israeli study of soldiers exposed to combat traumas10 suggested that prior combat stress reactions was a marker for subsequent similar problems rather than part of a process of aggregation of stress.
The majority of people will experience one or more traumatic events in their lifetimes, with estimates ranging from 51% to 90%11;12. Despite the shared nature of the experience, after natural disasters such as earthquakes, volcanic eruptions, tsunamis and of combat, only a minority of the population exposed to similar stressful experiences suffer from PTSD. Those who succumb are likely to have experienced prior psychiatric problems13;14;15. Prior psychiatric problems, less education, high neuroticism, extroversion, and certain ethnic grouping are associated with the development of PTSD 16;14;15;17;18;19;20;21;22. In the Blanchard et al17 study, prior depression was associated with Post Traumatic Stress Disorder following a motor vehicle accident to a highly significant extent, (P<0.004).
Risk factors
Pervasive factors which reconciles the various studies is the finding that temperamental characteristics, detected very soon after birth, proved to endure and to predict later behaviour and adjustment23. The personality dimension of adult neuroticism renders an individual vulnerable to neurotic disorders but there are elements of double counting. However, it is stable trait when corrected for age24. Those who are psychiatrically vulnerable are likely to succumb to the impact of significant life events, particularly if they have demonstrated that they have already done so in the past, which situation could be interpreted to indicate that they have less resilience to stress. Provocation tests are used in medicine, such as the glucose tolerance test for unmasking diabetes and the dexamethasone suppression test for testing for possible pituitary autonomous or semiautonomous malfunction in Cushing's syndrome. In an analogous fashion, it could be assumed that a latent psychiatric vulnerability would be revealed by psychosocial stress.
The propensity to experience traumatic events has cultural, educational and personality roots. Among young American adults, those with less education, blacks, and those with high extroversion scores (with a propensity for sensation seeking) are more likely thanothers to be exposed to traumatic events and are thus at greater risk forPTSD16. Age, gender, race, and socioeconomic status are relevant parameters, with youth, female sex and low socioeconomic status being markers for an increased likelihood of developing PTSD25;26;27;28. The level of social support and individual self-esteem, have also been implicated in the onset and course of PTSD across cultures29;30;31. This literature is suggestive of prior psychiatric, social and socio-economic pressures being conducive to PTSD and would inferentially support prior PTSD as being a marker, amongst other psychiatric markers, of psychiatric vulnerability arising from a variety of factors. A very recently published study from Naomi Breslau and her team, who have conducted a series of influential studies, brings additional and more direct evidence that prior PTSD is in fact a vulnerability marker, rather than the consequence of cumulative stress for the eruption of subsequent PTSD32. In this study, a sample of 1200 persons was randomly selected in 1989 from all 21-to 30-year-old members of a large health maintenance organization and were repeatedly assessed over a 10 year follow-up. The conditional risk of PTSD during the follow-up periods was found to be significantly higher among trauma-exposed persons who had experienced previous PTSD, relative to those with no prior trauma (odds ratio, 3.01; 95% confidence interval, 1.52-5.97). The estimates were only marginally revised after adjustment for sex, race, education, and pre-existing major depression and anxiety disorders. In contrast, the conditional risk of PTSD during follow-up among trauma-exposed persons who had experienced prior traumatic events but not PTSD was not significantly increased, relative to trauma-exposed persons with no prior trauma. The difference between the 2 estimates was significant (P = 0.005). The authors concluded that prior trauma increases the risk of PTSD after a subsequent trauma only among persons who developed PTSD in response to the prior trauma. The findings suggest that pre-existing susceptibility to a pathological response to stressors may account for the PTSD response to the prior trauma and the subsequent trauma.
One can only speculate as to why the individual developed PTSD in the first place. It has been argued on the basis of the Israeli combat data10 and natural disasters16;13;14;15 that the first episode of PTSD is likely to be a manifestation of psychiatric vulnerability rather than a manifestation of exposure to universally intolerable stress. These studies, particularly the most recent of Breslau and her team, have medico-legal implications which accord with the U.K. legal concepts of "nervous shock" and "eggshell personality" (Malcolm v Broadhurst [1970] 3 All ER 508).
In a similar fashion, Hammen et al33 found that non depressed persons were relatively resistant to the onset of depression, even when exposed to high- impact stressful events, whereas those who were symptomatic continued to have both more depression and more high-impact events over time. This may be partially a recording bias, because depressed people may better remember adversities and problems but there may be a further reason. In our competitive society, there seems to be some magnet-like effect of depressed people inducing others to assert their dominance and, metaphorically, to kick them while they are down, probably because of inadvertent signals of loss of self esteem and being an incapacitated adversary34. The converse is also true, with well-being decreasing life events vulnerability35. Brown Harris and Eales36 have illustrated the various implications of this interaction, particularly as to anxiety in the early and residual phases of depression. Recent biological evidence has shown a strong interconnection between the personality trait of neuroticism and depression. Neuroticism is associated with characteristics of serotonin receptors37;38 and the weight of evidence is that the short variant of the serotonin transporter gene is associated with depression39;40;41, although there is a contradictory study42.
Comorbidity
Between 48.6% and 51% of patients with a DSM-IIIR/DSM-IV diagnosis of major depression had at least one concomitant ('comorbid') anxiety disorder and only 26% to 34.8% had no comorbid mental disorder43;44. Comorbid depression occurred in 44.5% of PTSD patients at 1 month and in 43.2% at 4 months in 211 trauma survivors and was associated with greater symptom severity and lower levels of functioning13. In an Australian study, 21% of people fulfilling DSM-IV criteria for any mental disorder met the criteria for three or more comorbid disorders45. In a recent large-scale epidemiological study of 9,282 English-speaking respondents 18 years and older the researchers found that almost a quarter (23%) had 3 or more diagnoses, a situation which correlates with severity46. Using data from community surveys, many researchers have noted that if the range of psychiatric symptoms properly dictates 2 or more diagnoses, this is associated with greater symptom severity47;48;49;50;51, poorer outcome47;50 and poorer treatment response52, more functional impairment47,48;53 and increased use of medical service54;55;56;57 (see Wittchen58 for theoretical discussion of comobidity). Earlier, Kessler59 and Angst60 had noted that people who had more than one diagnosis at some time used services more often. Later work in a study of 10,641 adults showed a strong relationship between the number of disorders and disability and distress, with the combination of affective and anxiety disorders associated with four-fifths of the disability and service utilization45.
Ubiquity of stress
Certain stresses are inevitable, such as bereavement and one expects a period of readjustment. Despite the fact that stresses rain down upon us61;11;12;62 it is still the majority of the population who are not given a formal psychiatric diagnosis. A genetically determined constitution or an adverse childhood could well determine resilience and susceptibility to their impact. Complicating a model of exclusive social inculcation, childhood adversity is generally the product of parental behaviour by persons of shared genetic constitution.
Social Factors
The interaction between adversity and vulnerability, and the moderating effects of social integration and support are well known. Writing in the late 70s, a major research programme by Brown and Harris concluded: attention to a persons environment may turn out to be at least as effective as physical treatment." Clinically significant anxiety is much commoner amongst single people63. The situation is interactive in both directions. Psychiatric symptoms are likely to cause adverse social consequences and to be aggravated by these consequences, and multiple symptoms are more likely to have even greater adverse social resonances64;65;66;67;68;69;70;71.
A unifying model
The child is father of the man and half of all lifetime psychiatric cases start by age 14, and three fourths by age 24 46a, later-onset disorders occur in large part as temporally secondary comorbid conditions46b and the effect of aging on brain function may explain some of the late onset depressions72;73;74;75;76.
Linkage between the various neurotic disorders (i.e., anxiety, depressive, phobic, and obsessional neurosis) and the centrality of the neurotic disposition pervading all used to be commonly assumed. The subdivision in the DSM classificatory systems beginning with the third edition created a more rigid subdivision, which has been criticised adversely (e.g.77).
The personality factor of neuroticism is a pervasive risk factor for a variety of psychiatric illnesses (e.g.24;16;45). The temperamental characteristics may be no more than a forme fruste of illness which is highlighted by stress. People have varying coping capacities which are a product of their temperament and childhood experiences but the interaction, with adaptive and maladaptive responses, has even greater explanatory power. Parenting style and adequacy has been emphasised by the psychoanalytical movement and includes parental adjustment as interwoven with child rearing practices as well as with shared genetic propensities. On the other hand, enduring temperamental characteristics have been exhibited extremely early in life23;78;79 and are likely to be biologically determined and to introduce interactional biases in the developmental trajectory.
There are 3 models of the impact on mental health of adult trauma:
1. Repeated traumas, sometimes beginning in childhood produce a cumulative destructive effect and increasing sensitisation to further traumas (e.g.80).
2. Repeated small traumas produce a "stress inoculation" increasing resilience, in a manner analogous to the term battle hardened.
3. Prior psychiatric problems, perhaps surprisingly including PTSD, are markers of psychiatric vulnerability and predictors of subsequent psychiatric problems, including a further episode or episodes of PTSD.
The models do not conflict with one another but the recent study of Breslau et al32 provides empirical data emphasising the third possibility.
Treatment
Neurotic psychiatric disorders have a common underlying predisposing cause which can be modified by SSRIs and CBT. Claims are made for the effectiveness of exploring and reconciling childhood experiences and relationships and short-term psychodynamic psychotherapy has also proved to be an effective treatment (e.g.81). Depression is often mixed with anxiety or develops from anxiety. Anxietydisorders are by far the most common mental disorders but theproportion of serious cases is lower than for other classesof disorder. Mood disorders are the next most common with a higher proportion of serious cases46. Cognitive factors, especially the way people interpret or think about stressful events, are considered to play a pivotal role in the aetiology of anxiety82 and negative thoughts are frequently found in individuals with anxiety83. Anxiety is associated with a tendency to overestimate the association between a feared cue and personal harm84;85;86. Prominent negative thoughts in anxiety are underpinned by a sense of uncontrollability, feelings of helplessness and a perception that the sufferer is unable to predict, control, or obtain desired results82.
An amplifying factor in demands upon treatment is that high neuroticism contributes to patients' over-reporting of mood symptoms and help seeking, e.g.87. Appropriate treatment of neurotic disorders includes educating sufferers to identify and examine their negative thoughts, and to see if they can re-construe them in a more realistic and constructive way. Such cognitive behavioural therapy both promotes recovery and also guards against recurrence. In accord with biological findings regarding variance of the serotonin receptor, these twin advantages can also be obtained from continuing pharmacological treatments (e.g.88;67;89;90;91;92;93). The effect is amplified if the two approaches are applied in combination94;95;96;97.
ACKNOWLEDGEMENTS
I thank Dr. Martin Skelton-Robinson for helpful discussion.
COMPETING INTERESTS
I have prepared expert witness evidence where I have been instructed by claimants and defendants both jointly and severally
AUTHOR DETAILS
MALCOLM P WELLER, Honorary Research Professor, Middlesex University, UK
CORRESPONDENCE: MALCOLM P WELLER, Honorary Research Professor, Middlesex University, London NW4 4BT, UK
Email: psychiatry@weller.tv
References
Judd LL. Pleomorphic expressions of unipolar depressive disease: summary of the 1996 CINP President's Workshop. J Affect Disord. 1997; 45:109-116.
Andrews G. Should depression be managed as a chronic disease? Br Med J. 2001; 322:419-421.
Thornicroft G. and Sartorius N. The course and outcome of depression in different cultures: 10-year follow-up of the WHO Collaborative study on the assessment of depressive disorders. Psychol. Med. 1993; 3: 1023-1032.
Coryell W; Zimmerman M; Pfohl B. Short-term prognosis in primary and secondary major depression. J AFFECT DISORD. 1985; 9/3 265-270.
Smoller JW, Tsuang M. Panic and phobic anxiety: defining phenotypes for genetic studies. Am J Psychiatry. 1998; 155(9):1152-1162.
Lesser I.M., Rubin R.T., Pecknold J.C., et al. Secondary depression in panic disorder and agoraphobia, I: frequency, severity and response to treatment. Arch Gen Psychiatry. 1988; 45:437-443.
Bakker, A; Van Balkom, A; Spinhoven, P; Van Dyck, R. Follow-Up on the Treatment of Panic Disorder With or Without Agoraphobia: A Quantitative Review. J of Nerv and ment. Dis. 1998; 186(7) 414-419.
Rosenbaum JF, Pollack MH, Pollock RA. Clinical issues in the long-term treatment of panic disorder. J Clin Psychiatry. 1996; 57 (Suppl 10):44-48.
Symonds, Air Vice-Marshal Sir Charles P and Wing Commander Denis J Williams. Clinical and statistical study of neuroses precipitated by flying duties. Chap. 10 in Air Ministry Air Publication 3139, HMSO, London 1947.
Solomon Z, Mikulincer M, Jakob BR. Exposure to recurrent combat stress: combat stress reactions among Israeli soldiers in the Lebanon War. Psychol Med. 1987; 17(2):433-440.
Breslau N, Kessler R, Chilcoat HD, Schultz LR, Davis GC, Andreski P. Trauma and posttraumatic stress disorder in the community: The 1996 Detroit Area Survey of Trauma. Arch Gen Psychiatry. 1998; 55:626632.
Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995a; 52:1048-1060.
Shalev, A Y.; Freedman, S; Peri, T; Brandes, D; Sahar, T; Orr, S P.; Pitman, R K. Prospective Study of Posttraumatic Stress Disorder and Depression Following Trauma. Am J Psychiatry. 1998; Volume 155(5).May 630-637.
Sutherland, A G; Alexander, D A; Hutchison, James D. The Mind Does Matter: Psychological and Physical Recovery After Musculoskeletal Trauma. Journal of Trauma-Injury Infection & Critical Care. 2006; 61(6):1408-1414, December.
Zatzick DF, Kang SM, Muller HG, et al. Predicting posttraumatic distress in hospitalized trauma survivors with acute injuries. Am J Psychiatry. 2002; 159:941946.
Breslau N, Davis G C and Andreski P. Risk factors for PTSD-related traumatic events: a prospective analysis. Am J Psychiatry. 1995; 152:529-535
Blanchard, Edward B; Hickling, Edward J; Taylor, Ann E; Loos, W. Psychiatric morbidity associated with motor vehicle accidents. Journal of Nervous & Mental Disease. 1995; Vol 183(8) 495- 504.
North CS; Smith EM; Spitznagel EL. Post-traumatic stress disorder in survivors of a mass shooting. American J. Psychiatry. 1994; 151: 82-88.
Breslau N. Davis GC. Peterson EL. Schultz L. Psychiatric sequelae of posttraumatic stress disorder in women. Arch Gen Psychiatry. 1997; 54(1):81-7.
Blanchard E B and Hickling, E J. After The Crash: Assessment and Treatment of Motor Vehicle Accident Survivors, American Psychological Association (APA). 1997.
McFarlane A.C. The aetiology of post-traumatic morbidity: Predisposing, precipitating and perpetuating factors. British Journal of Psychiatry. 1989; 254: 221-228.
Tennant C; Streimer JH; Temperly H. Memories of Vietnam: Post-traumatic stress disorders in Australian veterans. Aust New Zealand J Psychiatry. 1990; 24: 29-36.
Thomas, A., Chess, S., and Birch, H.G. Temperament and behavior disorders in children. University of London Press. 1968.
Angst J. Course and prognosis of mood disorders 4.5.6, part of Chapter 4.5, 2000, - Mood disorders in: New Oxford Textbook of Psychiatry (1st Edition) Editors: Gelder, Michael G., Lopez-Ibor, Juan J., Andreasen, Nancy Publisher: Oxford University Press. 2000.
Brewin CR, Andrews B, Valentine JD. Meta-analysis of risk factors for posttraumatic stress disorder in trauma-exposed adults. J Consult Clin Psychol. 2000; 68:748-766.
Bromet EJ, Dew MA. Review of psychiatric epidemiologic research on disasters. Epidemiol Rev. 1995; 17:113-119.
Norris FH, Friedman MJ, Watson PJ, Byrne CM, Diaz E, Kaniasty K. 60,000 Disaster victims speak, part I: An empirical review of the empirical literature, 1981-2001. Psychiatry. 2002; 65:207-239.
Rubonis AV, Bickman L. Psychological impairment in the wake of disaster: The disaster-psychopathology relationship. Psychol Bull. 1991; 109:384-399.
Adams RE, Boscarino JA. Stress and well-being in the aftermath of the World Trade Center attack: The continuing effects of a community-wide disaster. J Commun Psychol. 2005; 33:175-190.
Boscarino JA. Post-traumatic stress and associated disorders among Vietnam veterans: The significance of combat exposure and social support. J Trauma Stress. 1995; 8:317-336.
Breslau N, Peterson EL, Schultz LR, Lucia VC. Estimating post-traumatic stress disorder in the community: Lifetime perspective and the impact of typical traumatic events. Psychol Med. 2004; 34:889-898.
Breslau N; Peterson E L; Schultz L R. A Second Look at Prior Trauma and the Posttraumatic Stress Disorder Effects of Subsequent Trauma: A Prospective Epidemiological Study. Arch Gen Psychiatry. 2008; 65(4):431-437.
Hammen C, Mayol A, deMayo R, Marks T. Initial symptom levels and the life-event-depression relationship. J Abnorm Psychol. 1986; 95:114-122.
Weller M P I and R. Priest R. Ethology In: The Scientific Basis of Psychiatry, Second edition, ed. M.P.I. Weller and M. Eysenck, W.B. Saunders, London, Philadelphia, Toronto etc. 1992.
Ryff CD, Singer B. The contours of positive human health. Psychol Inquiry. 1998; 9:1-28.
Brown GW, Harris TO, Eales MJ. Aetiology of anxiety and depressive disorders in an inner city population, 2: comorbidity and adversity. Psychol Med. 1993; 23:155-165.
Frokjaer V.G., Mortensen E.L., Nielsen F.A., et al. Frontolimbic serotoninc 2A receptor binding in healthy subjects is associated with personality risk factors for affective disorder. Biol. Psychiatry. 2008; 63: 569-576.
Takano A., Arakawa R., Hayashi M, Takahashi, H, Ito H, Suhara T. Relationship between enrutocisim personality trait and serotoninc transporter binding. Biol Psychiatry. 2007; Sep 15; 62(6): 588-92. Epub 2007 Mar 6.
Gonda, X, Rihmer, Z and Zsombok, T et al. The 5HTTLPR polymorphism of the serotonin transporter gene is associated with affective temperaments as measured by TEMPS-A, J. Affect. Disord. 2006; 91: 125-131.
Rihmer, Z, Gonda, X and Akiskal, K K et al. Affective temperament: a mediating variable between environment and clinical depression?, Arch. Gen. Psychiatry. 2007; 64: 1096-1097.
Schmitz, A, Hennig, J and Kuepper, Y et al., The association between neuroticism and the serotonin transporter polymorphism depends on structural differences between personality measures, Pers. Individ. Differ. 2007; 42:789-799.
Willis-Owen S.A.G. Turri. M.G, Munfa M.R., P.G. Surtees PG, N.W.J. Wainwright NW J , R.D. Brixey R D and J. Flintj, The serotonin transporter length polymorphism, neuroticism, and depression: a comprehensive assessment of association, Biol. Psychiatry. 2005; 58: 451-456.
Kessler, R. C., McGonagle, K. A., Zhao, S., et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States: results from the National Comorbidity Survey. Archives of General Psychiatry. 1994; 51: 8-19.
Wittchen, H.-U., Nelson, C. B. & Lachner, G. Prevalence of mental disorders and psychosocial impairments in adolescents and young adults. Psychological Medicine. 1998; 28: 109-126.
Andrews, G., Slade, T. & Issakidis, C. Deconstructing current comorbidity: data from the Australian National Survey of Mental Health and Well-Being. British Journal of Psychiatry. 2002; 181: 306-314.
46a. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age- of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry.2005; 62:593-602
46b. Kessler R C, Chiu W T , Demler O, Walters E E. Prevalence, Severity, and Comorbidity of 12-Month DSM-IV Disorders in the National Comorbidity Survey Replication Arch Gen Psychiatry. 2005; 62:617-627.
Clancy J., Noyes R., Hoenk P.R., et al. Secondary depression and anxiety neurosis. J.Nerv Ment Dis. 1978; 166:846-850.
Van Valkenburg C, Akiskal HS, Puzantian V, Rosenthal T. Anxious depression: clinical, family history and naturalistic outcome: comparisons with panic and major depressive disorders. J of Affective Disorders. 1984; 6: 67-82.
Vollrath, M. & Angst, J. Outcome of panic and depression in a seven-year follow-up: results of the Zurich study. Acta Psychiatrica Scandinavica. 1989; 80: 591-596.
Noyes, R., Reich, J., Christansen, J., et al. Outcome of panic disorder. Relationship to diagnostic subtypes and comorbidity. Archives of General Psychiatry. 1990; 447: 809-818.
Roy-Byrne, P., Vitaliano, P., Cowley, D., et al. Coping in panic and major depressive disorder. Relative effects of symptom severity and diagnostic comorbidity. Journal of Nervous and Mental Disease. 1992; 180: 179-183.
Keller, M. B., Lavori, P. W., Lewis, C. E. & Klerman, G. L. Predictors of relapse in major depressive disorder. Journal of the American Medical Association. 1983; 250: 3299-3304.
Roy-Byrne PP, Stang P, Wittchen HU, Ustun B, Walters EE. Lifetime panic-depression comorbidity in the National Comorbidity Survey. Association with symptoms, impairment, course and help- seeking. Br J Psychiatry. 2000,Mar; 176: 229-35.
Sturt, E. Hierarchical patterns in the distribution of psychiatric symptoms. Psychological Medicine. 1981; 11: 783-794.
Boyd, J. H., Burke, J. D., Gruenberg, E., et al. Exclusion criteria of DSM-III: a study of co-occurrence of hierarchy-free syndromes. Archives of General Psychiatry. 1984; 41: 983-989.
Andrews, G. Comorbidity and the general neurotic syndrome. British Journal of Psychiatry. 1996; 168 (suppl. 30): 76-84.
Kessler, R. C., Nelson, C. B., McGonagle, K. A., et al. Comorbidity of DSM-III-R major depressive disorder in the general population: results from the US National Comorbidity Survey. British Journal of Psychiatry. 1996; 168 (suppl. 30): 17-30.
Wittchen, H. U.Critical issues in the evaluation of comorbidity of psychiatric disorders. British Journal of Psychiatry. 1996; 168 (suppl. 30): 9-16.
Kessler, R. C. Epidemiology of psychiatric comorbidity. In Textbook in Psychiatric Epidemiology (eds M. T. Tsuang, M. Tohen & G. E. P. Zahner). 1995b: 179-198. New York: Wiley-Liss.
Angst J. Comorbidity of mood disorders: a longitudinal prospective study. Br J Psychiatry. 1996; Suppl Jun;(30):31-7.
Kendler KS, Kessler RC, Walters EE, et al. Stressful life events, genetic liability, and onset of an episode of major depression in women. Am J Psychiatry. 1995; 152:833-42.
Farmer AE, McGuffin P. Humiliation, loss and other types of life events and difficulties: a comparison of depressed subjects, healthy controls and their siblings. Psychological Medicine. 2003; 7: 1169-1175.
Reiger,D.A., Narrow, W.E. and Rae,D.S. The epidemiology of anxiety disorders: the epidemiology catchment area experience. J.Psychiat.Res. 1990; 24, Suppl.2: 3014.
Brown G W, Harris T. Social origins of depression. Tavistock Publications London. 1978.
Kedward H. The outcome of neurotic illness in the community. Social Psychiatry. 1969; 4: 1-4.
Figley C.R. Traumatic stress: the role of the family and social support system. In, Trauma and its Wake. The study of Post Traumatic Stress Disorder (ed. C.R. Figley), pp 39-56, Brunner/Mazel, New York. 1985.
Frank, E., Kupfer, D. J., Perel, J. M., et al. Three-year outcomes for maintenance therapies in recurrent depression. Archives of General Psychiatry. 1990; 47: 1093-1099.
Dalgard OS. Social support, negative life events and mental health. British Journal of Psychiatry. 1995; 166: 29-34.
King LA. King DW. Fairbank JA. Keane TM. Adams GA. Resilience-recovery factors in post-traumatic stress disorder among female and male Vietnam veterans: hardiness, postwar social support, and additional stressful life events. Journal of Personality & Social Psychology. 1998; 74(2):420-34.
George, L. K., Blazer, D. G., Hughes, D. C. & Fowler, N. Social support and the outcome of major depression. British Journal of Psychiatry. 1989; 154, 478-485.
Surtees, P. G. Social support, residual adversity and depressive outcome. Social Psychiatry. 1980; 15: 71-80.
Lockwood KA, Alexopoulos GS, van Gorp WG: Executive dysfunction in geriatric depression. Am J Psychiatry. 2002; 159:11191126.
Sanders M, Lyness JM, Eberly S, King DA, Caine ED: Cerebrovascular risk factors, executive dysfunction, and depression in older primary care patients. Am J Geriatr Psychiatry. 2006; 14:145152.
Alexopoulos GS, Meyers BS, Young RC, Kalayam B, Kakuma T, Gabrielle M, Sirey JA, Hull J: Executive dysfunction and long-term outcomes of geriatric depression. Arch Gen Psychiatry. 2000; 57:285290.
Alexopoulos GS: Role of executive function in late-life depression. J Clin Psychiatry. 2003; 64:1823.
Tyrer P, Seivewright H, Johnson T. The core elements of neurosis: mixed anxiety-depression (cothymia) and personality disorder. J Personal Disord. 2003,Apr; 17(2):129-38.
Chess, S. and Thomas, A. Origins and evolution of behavior disorders: from infancy to early adult life. BrunnerMazel, New York. 1984.
Thomas A, Chess S: Temperament and Development. Brunner-Mazel, New York. 1977.
Storr C L, Ialongo N S, Anthony J C, and Breslau N. Childhood Antecedents of Exposure to Traumatic Events and Posttraumatic Stress Disorder. Am J Psychiatry. 2007, January 1; 164(1): 119-125.
Leichsenring F., Rabung S., Leibing E. The Efficacy of Short-term Psychodynamic Psychotherapy in Specific Psychiatric Disorders. A Meta-analysis Arch Gen Psychiatry. 2004; 61:1208-1216.
Barlow, D. H., Chorpita, B. F., & Turovsky, J. Fear, panic, anxiety, and disorders of emotion. In D. A. Hope (Ed.), Perspectives on anxiety, panic, and fear. The 43rd Annual Nebraska Symposium on Motivation, pp. 251-328. Lincoln, NE: Nebraska University Press. 1996.
Ingram RE, Miranda J, Segal ZV: Cognitive Vulnerability to Depression. New York, Guilford Press. 1998.
Tomarken, A.J., Davidson, R.J., and Henriques, J.B. Resting frontal brain asymmetry predicts affective responses to films. Journal of Personality and Social Psychology. 1990; 59: 791-801.
de Jong, P.J., Merckelbach, H., and Arntz, A. Covariation bias in phobic women: the relationship between a priori expectancy, on-line expectancy, autonomic responding, and a posteriori contingency judgement. Journal of Abnormal Psychology. 1995a; 104: 55-62.
de Jong, P.J., van den Hout, M.A., and Merckelbach, H. Covariation bias and the return of fear. Behaviour Research and Therapy. 1995b; 33: 211-13.
Duberstein P R and Heisel M J. Personality traits and the reporting of affective disorder symptoms in depressed patients. Journal of Affective Disorders. 2007, Nov; 103 (1-3): 165-171.
Fog R; Lader M; Montgomery S. 'Citalopram - a well- documented and novel antidepressant' Int Clin Psychopharmacol. International Clinical Psychopharmacology. 1995; 10/SUPPL. 1 (3).
Prien, R. F. & Kupfer, D. J. Continuation drug therapy for major depressive episodes: how long should it be maintained? American Journal of Psychiatry. 1986; 143: 18-23.
Melfi, C. A., Chawla A. J., Croghan, T. W., et al. The effects of adherence to antidepressant treatment guidelines on relapse and recurrence of depression. Archives of General Psychiatry. 1998; 55: 1128-1132.
Maj, M., Veltro, F., Pirozzi, R., et al. Pattern of recurrence of illness after recovery from an episode of major depression: a prospective study. American Journal of Psychiatry. 1992; 149: 795-800.
Claxton A. J. and McKendrick J. Selective serotonin reuptake inhibitor treatment in the UK: risk of relapse or recurrence of depression. The British Journal of Psychiatry. 2000; 177: 163-168.
Segal, Z., Pearson, J. & Thase, M. E. Challenges in preventing relapse in major depression. Report of a National Institute of Mental Health Workshop on state of the science of relapse prevention in major depression. Journal of Affective Disorders. 2007; 77: 97-108.
Otto MW, Hinton D, Korbly NB, Chea A, Phalnarith B, Gershuny BS, Pollack MH. Treatment of pharmacotherapy-refractory posttraumatic stress disorder among Cambodian refugees: a pilot study of combination treatment with cognitive-behavior therapy vs sertraline alone. Behav Res Ther. 2003; 41: 12711276.
Keller MB, McCullough JP, Klein DN, Arnow B, Dunner DL, Gelenberg AJ, Markowitz JC, Nemeroff CB, Russell JM, Thase ME, Madhukar H, Trivedi MH, Zajecka J. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med. 2000; 342: 14621470.
Thase ME, Greenhouse JB, Frank E, Reynolds CF III, Pilkonis PA, Hurley K, Grochocinski V, Kupfer DJ: Treatment of major depression with psychotherapy or psychotherapy-pharmacotherapy combinations. Arch Gen Psychiatry. 1997; 54: 10091015.
Pampallona S, Bollini P, Tibaldi G, Kupelnick B, Munizza C: Combined pharmacotherapy and psychological treatment for depression: a systematic review. Arch Gen Psychiatry. 2004; 61: 714719.
Bacterial infections are associated with many autoimmune diseases involving chronic inflammation and demyelination. The possible mechanisms of bacterial involvement as aetiological agents or in the exacerbation of these diseases have been investigated intensively. This review focuses the role of bacterial infections in the pathogenesis of autoimmune, inflammatory and demyelinating diseases. Possible modes of pathogenic action of bacteria are discussed, viz. the role of cytokines, Toll-like receptor signalling, the interaction of heat shock proteins with the immune system, and the role of nitric oxide. An auto-regulatory loop might exist in the interaction of bacteria with the host and in pathogenic signal processing. These studies reveal potential therapeutic targets.
Abbreviations: AQP4 Aquaporin-4; AS ankylosing spondylitis; CSF cerebrospinal fluid; EAE autoimmune encephalomyelitis; GB Guillain-Barre syndrome; HLA human leukocyte antigens; HSP heat shock protein; IL interleukin; LPS lipopolysaccharides; MAM Mycoplasma arthritidis antigen; MHC [proteins encoded by] major histocompatibility gene complex; MS multiple sclerosis; NK natural killer cells; NMO neuromyelitis optica; NO nitric oxide; NOS nitirc oxide synthase; PCR polymerase chain reaction; RA rheumatoid arthritis; SLE systemic lupus erythematosus; TLR Toll-like receptors; TNF tumour necrosis factor
Introduction
Bacterial and viral infections are commonplace in a variety of autoimmune and chronic illnesses such as the chronic fatigue syndrome (myalgic encephalomyelitis), fibromyalgia syndrome, Gulf War illnesses and rheumatoid conditions1-3. Much attention is focused at present on the role of bacteria and the possible mechanisms of their involvement in the pathogenesis of several diseases. The route of infection and penetration and the immune responses of the host can not only make any bacterial infection pathogenic but probably can also determine the aggressiveness of the disease and the chance for full recovery. Therefore the two basic elements addressed here are the association between bacterial infection and autoimmune disease and the involvement of the immune system in the disease process.
Bacterial infections in rheumatoid conditions
A wide variety of bacterial infections have been associated with rheumatoid conditions. Rheumatic diseases might have a manifold aetiology with varying genetic susceptibility, but bacteria-related autoimmunity might be an important factor4.
Mycoplasma infection, e.g. by M. pneumoniae, M. salivarum, and M. fermentans, has been strongly associated with RA (rheumatoid arthritis)5-8. There is often systemic infection of more than one species8. Mycoplasma antigens induce both cell-mediated and humoral immune responses. Enhanced levels of antibodies against MAM (Mycoplasma arthritidis antigen) have been found in sera from RA patients in comparison with antibodies against Staphylococcal enterotoxins A and B. Also antibody titers were higher in RA serum than in systemic lupus erythematosus, ankylosing spondylitis, psoriatic arthritis, Reiter's syndrome, or healthy controls.
The mycoplasma antigen MAM can activate T cells. MAM contains two domains, one of which can inhibit lymphocyte proliferation; the second domain, which contains concanavalin A motif-, is required for T cell activation9. It can also up regulate natural killer cell activity10. Furthermore, synovial tissues of RA patients contain T-cells, which bear the same T-cell receptors as used by MAM. The mitogen seems to be capable of initiating and exacerbating arthritic changes11, 12. MAM is a zinc-dependent antigen that binds to MHC class II molecules. Zinc induces MHC protein dimerisation required for MAM binding, MHC-induced cell-cell adhesion, and efficient T cell activation13, 14. As discussed in later sections, MAM can alter cytokine expression profiles and activate and modulate nitric oxide synthase (NOS) signalling pathways.
Bacterial DNA isolated from rheumatoid arthritis (RA) and juvenile arthritis has included Haemophilus influenzae, Bordatella and Yersinia as possible infecting organisms15. Lyme arthritis, which resembles rheumatoid synovial infiltration by Borrelia burgdorferi, has often been suggested to be an autoimmune condition. The B. burgdorferi surface protein A (OspA161-175) is recognised by T-cells and HLA (human leukocyte antigen)-DR molecules that bind this T-cell epitope and to these events is attributed the development of autoimmunity following B. burgdorferi infection. However, these decline with antibiotic therapy16. Therefore, in spite of the perceived association, Drouin et al.17 diligently searched for peptides with sequence homology with OspA (165-173) and have concluded from their study that molecular mimicry might not be significant to pathogenesis. The epitope OspA (163-175) is the predominant epitope associated with Lyme disease. Serum reactivity against OspA is also found in RA patients18. Our knowledge concerning the interaction of B. burgdorferi with host tissues and cells is rather scant. Ghosh et al.19 have suggested cytokeratin 10 as a potential autoantigen. Gavanescu et al.20 reported that mycoplasma infections can result in the production of autoantibodies against centrosomes. It is not known if this cellular organelle is involved with autoimmunity in RA.
B. burgdorferi seems to be able to induce inflammatory responses including secretion of cytokines and cellular responses of the T-helper cell-1 (Th-1) type21. Beermann et al.22 generated lipoprotein vesicles (LV) from this bacterium and incorporated them into peripheral blood mononuclear cells. The resultant LV-T cells were predominantly the immune effector CD8+. Furthermore, these cells destroyed autologous T-cells carrying LV. These data do indeed support the existence of an autoimmune condition. Overall, a conservative conclusion would be that the molecular mimicry and autoimmunity thesis is yet to be fully tested.
Proteus mirabilis has been implicated in the pathogenesis of RA23-26 and in osteoarthritis (OA)27, 28. Again, the HLA DRB1 alleles appear to be the major genetic susceptibility factors as postulated some years ago29.
Bacterial infection associated with other autoimmune conditions
Bacterial infections have been identified in association with other autoimmune conditions besides RA. Members of the Enterobacteriaceae family are associated with autoimmune conditions such as Kawasaki syndrome and Graves disease. Demyelinating diseases have been a focus of active investigation in the past few years. Kollef et al.30 suggested that central and peripheral nerve demyelination might occur following M. pneumoniae infection. Since then patients with the autoimmune condition SLE (systemic lupus erythematosus) have been investigated for mycoplasma infections. Early studies revealed differences between SLE patients and control subjects in respect of genitourinary mycoplasma infections31. However, the deployment of more sensitive methods of detection has not supported these early claims. Runge et al.32, for instance, found no difference in the incidence of Ureaplasma urealyticum in SLE patients, and they discount the notion that this mycoplasma species has any role to play in the pathogenesis of SLE. Nonetheless, there should be no serious doubts that mycoplasma infection can lead to demyelination.
The demyelinating neuropathy known as Guillain-Barre (GB) syndrome often has pathogenic association with bacterial infections. Campylobacter jejuni, Haemophilus influenzae and M pneumoniae have been implicated as possible causative agents of GB. C. jejuni is the major infecting organism here together with M. pneumoniae infection in some cases33, 34. GB is associated with the presence of antibodies against galactocerebroside, which is a major component of myelin35, 36. Some bacterial LPS (lipopolysaccharides) apparently bear molecular similarity to the human gangliosides GM1, GM1b, GD1a, and GalNAc-GD1a of the motor axolemma and are said to be the target epitopes for antibodies occurring in the GB subtype acute motor axonal neuropathy. The antiganglioside antibodies cause axonal neuropathy37. The host immune response to LPS moieties of the HB93-13 strains of C. jejuni cross-reacts with human nerve gangliosides and induce GBS38. .
Multiple sclerosis (MS) is a chronic inflammatory and demyelinating disease of the central nervous system. The pathogenesis of MS is possibly a consequence of autoimmune condition or infection by viral or bacterial agents. Both infections lead to the development of demyelinating plaques. Bacterial infections can evoke immune responses and induce demyelination. Infections of the brain parenchyma are sequestered from the immune system. Matyszak39 has postulated a loss of the integrity of the blood-brain barrier at the foci of infection by a delayed-type hypersensitivity response leading to demyelination. Nitric oxide (NO), which enhances the permeability of the blood brain barrier, is found in greater quantities in the CSF (cerebrospinal fluid) of MS patients than of control subjects4. Also NO metabolite levels reportedly correlate with disease activity41. Other explanations have also been advanced. Gay42 has drawn attention to the putative link of bacterial nasopharyngeal infections with optic neuritis, optochiasmatic arachnoiditis and MS. The possibility is aired that the blood barrier may be by-passed. Gay42 points out the physical connection between CSF and the lymphatic drainage channels of the nasopharyngeal mucosa. So in the event, the CNS could be exposed to bacterial toxins and generate an immunological response. Many autoimmune diseases involve HLAs. The latter play a key role in antigen presentation to CD4+ Th cells. Specific regions of HLA e.g. HLA-C have robust association with MS and Graves' disease43.
More recently serology and PCR (polymerase chain reaction) have provided ample evidence of Chlamydia pneumoniae, Borrelia burgdorferi, Mycoplasma species, human herpesvirus-1 and -6, among others in MS, amyotrophic lateral sclerosis Alzheimer's and Parkinson's disease3. Parratt et al.44 looked at Chlamydia pneumoniae-specific immune complexes and have reported that C. pneumoniae infection is more frequent in MS patients and detected early in the course of the disease, presumably indicating an aetiological link. A tentative relationship between MS and streptococcal infection has been suggested45. But Budak et al.46 have found no evidence of C. pneumoniae DNA in CSF samples. Similarly no Mycoplasma-specific nucleic acid sequences were detected47 (Casserly et al. 2007). Lindsey and Patel48 found no trace of bacterial 16S DNA in the CSF of MS patients with progressive diseases or patients in remission. They tested for Campylobacter, Mycoplasma, Chlamydia, Bartonella, Mycobacteria and Streptococcus.
The aetiology and pathogenesis of MS and neuromyelitis optica (NMO) have been studied intensively from another angle in recent years. MS and NMO are related conditions; NMO could be a variant of greater severity than MS. Specific autoantibody responses have been identified in NMO patients. These are AQP4 (Aquaporin-4) antibodies generated against the water channel protein AQP449. 50 and they selectively target astrocytic end feet at the glia limitans (the external glial limiting membrane).
AQP4 antibodies are regarded as autoantibodies and so associated with the pathogenesis of NMO. The occurrence of lesions in the brain and spinal cord of NMO patients are consistent with the degree of AQP-4 expression51. The damage of astrocytes encountered in NMO is attributed to these antibodies. Serum IgG from patients with NMO binds to the extracellular domain of aquaporin-4; it is predominantly IgG1. Antibody binding to membranes expressing aquaporin-4 probably initiates demyelination52. HSP (heat shock protein) 70 has also been implicated in the autoimmune response leading to demyelination53.
Possible modes of pathogenic action of bacteria
Molecular mimicry in pathogenesis
Most autoimmune diseases are associated with HLA types. RA and AS (ankylosing spondylitis) are classical examples of the association of HLA with rheumatoid conditions. More than 90% of RA patients possess HLA-DR1 or other sub-type and >96% of AS patients reportedly possess HLA-B2754. HLA-B27 antigen is also involved in reactive arthritis55. Spondyloarthropathies (SAP) are a group of HLA-B27-linked diseases, characterised by inflammatory pain in the spine and asymmetrical arthritis in the lower limbs. HLA-B27 transgenic mice spontaneously develop arthritis and they are susceptible to collagen-induced arthritis56. The involvement of HLA antigens in the pathogenesis of autoimmune diseases has been suggested to be due to the molecular similarities between certain bacterial antigens and HLA antigens. But there are no cross-reactive antibodies against bacteria and HLA-B27 in significant titres57.
The autoimmune conditions of Graves disease and Kawasaki syndrome are a result of the hyperactivation of the immune system. Bacterial infections have been implicated in both disease states. Yersinia enterocolitica, a member of the Enterobacteriaceae family, produces lipoproteins that are well known for their mimicry of the extracellular domain of the human thyrotropin receptor protein58, 59. This lipoprotein is mitogenic to B-cells60 and induces the production of autoantibodies against the thyrotropin receptor. This could be the cause of hyperthyroidism associated with Graves disease.
Chlamydial infection occurs commonly in chronic and acute diseases of the upper and lower respiratory tract and. also with atherosclerosis and asthma. Rheumatic autoimmune conditions also often show antibodies against the ribosomal protein L7. The L7 protein has been reported to contain epitopes bearing homology with a specific aminoacid sequence of the C. trachomatis RNA polymerase61. This has led to the suggestion that certain rheumatoid conditions could be due to the molecular similarity between L7 and the homologous sequence of the polymerase and generation of autoantibodies.
The autoimmune condition of SLE causes glomerulonephritis, arthritic changes and neurological alterations. SLE is another example of pathogenesis attributable to molecular mimicry between antigens of infecting agents and autologous proteins. As stated earlier, Campylobacter jejuni is the major infecting agent in patients with this disease. Hughes et al.62 demonstrated the presence of antibodies against the ganglioside GM1 in SLE patients. Campylobacter infection has been linked with the perceived molecular mimicry of bacterial LPS with the human gangliosides of the motor axolemma and these are the target epitopes for antibodies that are believed cause axonal neuropathy37.
The role of cytokines in the pathogenesis of autoimmune conditions
Another line of evidence that links bacterial infections with RA and other inflammatory autoimmune conditions is the demonstration that bacterial antigens, such as MAM, induce the synthesis of pro-inflammatory cytokines, interleukin (IL)-1, IL-6, and IL-863, 64. Furthermore, the modulation of the synthesis of cytokines by MAM corresponds with the induction of arthritic changes in mice65. The induction of IL-13 expression appears to be up regulated in human fibroblast cell lines when the cell cultures are contaminated by mycoplasmas66. IL-6 and IL-8 are induced in human gingival fibroblasts by a host of mycoplasma species, e.g. M. hominis, M. arthritidis, M. arginini, M. fermentans, M. penetrans, M. pirum and M. pneumoniae64. Glycolipid antigens of M. fermentans have been identified as important mediators of pathogenicity. The induction of TNF (tumour necrosis factor) together with cytokines and prostaglandins was reported some years ago67, 68. TNF is produced in response to M. fermentans antigens69. TNF- induced by M. fermentans appears to enhance cytokines that can modulate the immune system. Exposure of human lung fibroblasts to M. fermentans induces IL-6, IL-10 and IL-12, IL-1b, IL-8 (now designated as CXCL8), the monocyte chemoattractant protein-1 (MCP-1) also known as CCL2 (CC chemokine ligand 2), and the chemokine (C-X-C motif) ligand 1 (CXCL1) production69, 70. Kawahito et al.71 used monoclonal antibody against the M. fermentans glycolipid antigen GGPL-III and detected it in synovial tissues from RA patients. The antigen was not detectable in OA or normal synovial tissues. Furthermore, GGPL-III induced TNF-a and IL-6 production by peripheral blood mononuclear cells, and also induced proliferation of synovial fibroblasts. However, anti-phospholipid antibodies are generated in response to a number of bacterial infections72. So their presence alone might not be clearly linked with pathogenesis.
Toll-like receptor signalling in immune responses to infection
The generation of immune responses to lipopolysaccharides (LPS) is mediated by a class of receptors called Toll-like receptors (TLRs). These are transmembrane receptors that activate immune cell responses. TLR can recognise molecular patterns associated with pathogenic infectious agents; among them of note are LPS, viral RNA, and unmethylated CpG-oligonucleotides.
The TLR signalling mechanism has been the focus of much attention. Exposure of cells to LPS or other toxins induces the expression of different forms of TLR and different pro-inflammatory interleukins and interferons73. Inflammatory responses to M. arthritidis lipoproteins require TLRs74. MAM of M. arthritidis has been shown to interact with TLRs75. MAM generates a differential immune reactivity mediated by different TLR types. The co-stimulatory molecules associated with the immune stimulation determine the outcome in terms of IL isoform produced and this depends upon which co-stimulatory factor interacts with MAM. Thus inhibition of co-stimulatory factor B7-1 leads to a shift from IL-2 to IL-176. TLR signalling also involves caspases required for processing the precursors of IL-1 and IL-18. The TLRs use the adapter protein MyD88 and the so-called adapter-like MyD88 to activate signaling pathways, but only the latter interacts with caspase77. So here we have another potential means of regulating the expression pattern of pro-inflammatory cytokines. In other words, IL production pattern is determined by the co-operation TLRs. TLRs can synergistically or competitively modulate IL expression in immune response to infectious agents78. Equally, one can attribute specific TLRs of T-cells with the ability to directly stimulate Th1 and Th2 effector function and modulate the synthesis of cytokines and interferons, and influence cell proliferation and survival79. TLR function is closely related to Fc? receptor (Fc?R) expression. The cells of the immune system express receptors for the Fc region of Ig isotypes. Fc?R for IgG links IgG mediated responses of the immune system80. TLR4 up regulates the expression of Fc?R. IL10 is said to be involved in and mediate this up regulation81. Probably, as Loof et al.82 have implied, TLRs might be functioning as a cohort of signalling channels interacting with one another rather than acting individually to generate an immune outcome. TLR signalling might be autoregulated; a concept that is worthy of investigation.
LPS seems to induce the production of interleukins via a TLR-mediated pathway. Exposure of the macrophage RAW264.7 cell line to LPS leads to Janus kinase (JAK)2 tyrosine phosphorylation with TLR4 mediation, then down stream to the phosphorylation of JNK {c-jun N terminal kinase) resulting in IL production83.
Finally, TLR signalling is involved in the activation of innate immunity in defence from infections not only bacterial, but also viral and parasitic. NK cells, macrophages, dendritic cells are all capable of enlisting TLRs signalling in their function. The recognition of bacterial infection by NK cells seems to be mediated by TLR84, 85. Other infections e. g. by the parasitic protozoan Leishmania major, result in the induction of IL-12 in bone marrow-derived dendritic cells, IFN-? expression and activation of NK cells. These events are mediated by TLR986.
The interaction of heat shock proteins with the immune system
Heat shock proteins (HSP) are a highly conserved family of stress-related proteins with diverse function such as protein folding and chaperoning, and novel and differential modes of function have now been ascribed to their functional repertoire. HSPs might chaperone antigenic peptides.
Antibodies against a number of HSPs have been detected in autoimmune diseases. Marked increases in antibodies against HSP70 and HSP90 occur in patients with RA87, Klebsiella pneumoniae HSP60 in ankylosing spondylitis patients88, HSP27 and HSP90 antibodies in patients with arthritis accompanying cystic fibrosis89 and so on. T lymphocytes react to heat shock proteins and this probably plays an important regulatory role in the progression of autoimmune diseases. HLA-DR-T cell epitopes have been identified in HSP60 and HSP7090-92. In experimental systems HSP60 induces the production of IL-8 and TNF (tumor necrosis factor)-a and this is enhanced by HSP auto-antibodies. Sera from RA patients with higher anti-HSP60 auto-antibody titers also markedly increased the IL-8 production induced by HSP60 in a human monocytic cell line93. As yet, it is unclear what role HSP auto-antibodies might play in pathogenesis.
HSP are known to be able to influence both innate and adaptive immune response and induce the expression of interleukins under a variety of experimental conditions. Some HSPs induce and other can inhibit the production of interleukins. Bacterial HSPs bear sequence homology to human HSPs, and immunisation with bacterial HSPs has often inhibited disease progression94. Several HSP receptors have been identified to-date on antigen presenting cells. Among them are the Toll-like receptors TLR2 and TLR4. HSPs are recognised by appropriate receptors to initiate their participation in the signalling cascade95, 96. Singh et al.97 showed that heat shock activated transcription factor HSF-1 (heat shock factor-1) binds to heat shock responsive elements in the promoter of genes coding for certain interleukins.
The TLR signalling pathway has been robustly implicated in HSP function. HSPs recognise and bind to pathogen-associated molecules and activate TLR mediated signalling. It appears possible that different HSPs might differentially activate TLRs thus determining the functional pathway. Thus HSP60 is said to bind to TLR1 but not to TLR298; this could have differing consequences in terms of induction of cytokines. HSP70 bound to the cell membrane is said to specifically activate NK cells, whilst intracellular HSP70 exerts immunomodulatory effects by binding specifically to TLR2 and TLR4. In vitro studies have suggested that HSP70 is actively released in response to heat shock and induces the production of IL-10 in fibroblast-like synoviocytes by TLR4 signalling99. HSP72 induces IL-8 expression by activating TLR4 and NF-kappa B100.
The role of nitric oxide in the pathogenesis of autoimmune, inflammatory and demyelinating diseases
Nitric oxide (NO) is synthesised by NO synthase (NOS) which occurs as neuronal, endothelial and inducible isoforms. NO subserves many functions. Most prominently, it is a vasoactive agent regarded as contributing significantly to the pathogenesis of inflammatory immune and neurodegenerative diseases. RA, SLE, MS, and experimental autoimmune encephalomyelitis (EAE), an experimental model of MS, all show associated synthesis of NO, superoxide and their toxic products. Upon infection bacterial components bind to macrophages using TLRs and this leads to the production of TNF-a, which in turn induces to the synthesis of NO. NO is also expressed by cells when exposed to IFN-?. NOS is required for bacterial clearance during infection101. The general and overall effects would be bactericidal in nature.
Arthritic changes occurring in an animal model called adjuvant-induced arthritis, which exhibits features similar to those of RA, accompany the induction of NOS. Furthermore, NOS inhibitors suppress the arthritic changes102. Mouse peritoneal macrophages and a macrophage cell line have been reported to synthesise NO in response to MAM. This is enhanced by LPS possibly via TLR2 but not TLR4 signalling103, 104. M. hominis lipophilic component also interacts with TLR2 not TLR4105. M. synoviae lipoprotein lipid moiety induces NO secretion by chicken macrophages106.
Nitric oxide (NO) enhances the permeability of the blood brain barrier. The invasion of the CNS by inflammatory cells and the development of EAE are prevented if the toxic product peroxynitrite of NO and superoxide are scavenged107. Both constitutive isoforms of NOS, neuronal and endothelial, and inducible NOS are active in the demyelination process108. NOS has also been implicated in the pathogenesis of Parkinsons disease109.
Demyelination can be induced by mycoplasmas. NO, inflammatory cytokines, and prostaglandins are induced when glial cells are exposed to M. fermentans antigens68. Heat shock inhibits both NO and iNOS (inducible NOS)110, 111. Bacterial LPS-induced expression of NOS can be inhibited by exposure of cells to hyperthermia at 43C. Transfection of HSP70 reduces iNOS expression111, 112. From the foregoing discussions one can visualize a complete regulatory picture of the involvement of HLAs, HSPs, cytokines and nitric oxide in the pathogenesis of inflammatory immune diseases.
Although NO is harmful to bacteria, it can induce apoptosis in some cell systems113 and cause necrosis (see Naito et al.114). The toxicity of NO can result in immune suppression and in turn lead to enhanced infection115. Bacteria also seem to have evolved protective mechanisms against these deleterious effects. So host resistance to bacterial infections and the ability of bacteria to initiate inflammatory and demyelinating conditions leading to pathogenesis are finely tuned. The understanding of the control mechanisms has not only expanded our knowledge of the possible modes of bacterial involvement in pathogenesis, but it has also led to the identification of potential targets for therapy. The activation of signalling pathways mediated by TLRs has afforded an avenue of therapeutic approaches to autoimmune conditions. TLR signalling together with its cognate receptors and adapter molecules can conceivably be employed as specific targets for therapeutic intervention. The TLR agonists have been found to enhance immune responses, especially against tumours116. Some agonists have been approved for the treatment of certain human disease conditions117, 118. There is also much scope for the detection of bacterial infections via TLRs. As discussed earlier the TLR signalling pathway might be implicated in potential crosstalk with other interacting signalling systems. In other words, a composite regulatory operation of many pathways involving TLRs can be delineated in the pathogenesis of autoimmune diseases.
It would not be out of place to inquire here into the potential clinical benefits of studying the role of bacterial infections and the mode of their participation in the disease process. The prevention of disease progression is one of the benefits, in which not only the identification of the infecting agent but also the mode by which the infectious agents might trigger initiation and progression would make a valuable contribution. Specifically targeted intervention modalities using antibacterial therapy can evolve and develop from such basic research. Also these would find much application in the development of healthcare facilities such as antimicrobial stewardship programmes and infection control programmes to monitor effects of treatment and treatment costs119. Unavoidably the cost effectiveness of treatment regimes comes into reckoning. This makes it imperative that factors which determine antibiotic-resistance of bacteria are identified and adequately addressed.
ACKNOWLEDGEMENTS
I thank Professor Jrg Haier of the Cancer Centre of the University Hospital at Mnster and Dr M.S. Lakshmi for reading the manuscript and making valuable suggestions, and Professor Bayan Sharif and Professor Satnam Dlay for supporting my research and literary efforts.
COMPETING INTERESTS
None Declared
AUTHOR DETAILS
GAJANAN V SHERBET, The Institute for Molecular Medicine, Huntington Beach CA, USA
CORRESPONDENCE: GAJANAN V SHERBET, School of Electrical, Electronic and Computer Engineering, University of Newcastle upon Tyne, Merz Court, Newcastle upon Tyne, NE1 7RU, U.K.
Email: gajanan.sherbet@ncl.ac.uk
References:
1. Nicolson GL, Nasralla M, Haier J, et al. Diagnosis and treatment of mycoplasmal infections in fibromyalgia and chronic fatigue syndromes. Relationship to Gulf war illness. Biomed Therapy 1998; 16: 266-271.
2. Nicolson GL, Nasralla MY, Franco AR, et al. Role of mycoplasmal infections in fatigue illnesses. Chronic fatigue and fibromyalgia syndromes, Gulf War illness and rheumatoid arthritis. J Chronic Fatigue Syndrome (Microbiol) 2000; 6: 23-29.
3. Nicolson, GL. Chronic bacterial and viral infections in neurodegenerative and neurobehavioral diseases. Labmed 2008; 39: 291-299.
5. Furr PM, Taylor-Robinson D, Webster ADR. Mycoplasmas and ureaplasmas in patients with hypogammaglobulinemia and their role in arthritis: Microbiological observations over twenty years. Ann Rheum disease 1994; 53: 183-187.
6. Simecka JW, Rosss SE, Cassell GH, et al. Interactions of mycoplasmas with B cells. Production of antibodies and nonspecific effects. Clin Infectious Dis 1993; 17: S176-S182.
7. Hoffman RW, OSullivan FX, Schafermeyer KR, et al. Mycoplasma infection and rheumatoid arthritis. Analysis of their relationship using immunoblotting and an ultrasensitive polymerase chain reaction detection method. Arthritis Rheum 1997: 40: 1219-1228.
8. Haier J, Nasralla M, Franco AR, et al. Detection of mycoplasmal infections in blood of patients with rheumatoid arthritis. Rheumatol 1999; 38: 504-509.
9. Cole BC, Knudtson KL, Oliphant A, et al. The sequence of the Mycoplasma arthritidis superantigen, MAM. Identification of functional domains and comparison with microbial superantigens and plant lectin mitogens. J Exp Med 1996; 183: 1105-1110.
10. DOrazio JA, Cole BC, Stein-Streilein J. Mycoplasma arthritidis mitogen up regulates human NK cell activity. Infection Immunity 1996; 64: 441-447.
11. Cole BC, Griffiths MM. Triggering and exacerbation of autoimmune arthritis by the mycoplasma arthritidis superantigen MAM. Arthritis Rheum 1993; 36: 994-1002.
12. Cole BC, Sawitzke AD, Mu HH. Mycoplasma arthritidis and its superantigen M-arthritidis mitogen as a model for inflammatory and autoimmune disease. EFFECTS Mcrobes Immune Sys 2000; 93-107.
13. Langlois MA, El Fakhry Y, Mourad W. Zinc-binding sites in the N terminus of Mycoplasma arthritidis-derived mitogen permit the dimer formation required for high affinity binding to HLA-DR and for T cell activation. J Biol Chem 2003; 278: 22309-22315.
14. Li HM, Zhao YW, Guo Y, et al. Zinc induces dimerization of the class II major histocompatibility complex molecule that leads to cooperative binding to a superantigen. J Biol Chem 2007; 282: 5991-6000.
15. Wilkinson NZ, Kindsley GH, Jones HW, et al. The detection of DNA from a range of bacterial species in the joints of patients with a variety of arthritides using a nested, broad-range polymerase chain reaction. Rheumatology 1999; 38: 260-266.
16. Kannian P, Drouin EE, Glickstein L, et al. Decline in the frequencies of Borrelia burgdorferi OspA(161-175)-Specific T cells after antibiotic therapy inHLA-DRB1*0401-positive patients with antibiotic-responsive or antibiotic-refractory Lyme arthritis. J Immunol 2007; 179: 6336-6342.
17. Drouin EE, Glickstein L, Kwok WW, et al. Human homologues of a Borrelia T- cell epitope associated with antibiotic-refractory Lyme arthritis. Mol Immunol 2008; 45: 180-189.
18. Hsieh YF, Liu HW, Hsu TC, et al. Serum reactivity against Borrelia burgdorferi OspA in patients with rheumatoid arthritis. Clin Vaccine Immunol 2007; 14: 1437-1441.
19. Ghosh S, Seward R, Costello CE, et al. Autoantibodies from synovial lesions in chronic, antibiotic treatment-resistant Lyme arthritis bind cytokeratin-10. J Immunol 2006; 177: 2486-2494.
20. Gavanescu I; Pihan G, Halilovic E, et al. Mycoplasma infection induces a scleroderma-like centrosome autoantibody response in mice. Clin Exptl Immunol 2004; 137:288-297.
21. Franz JK, Priem S, Rittig MG, et al. Studies on the pathogenesis and treatment of Lyme arthritis. Wiener Klini Wochenschrift 1999; 111, 981-984.
22. Beermann C, Wunderli-Allenspach H, Groscurth P, et al. Lipoproteins from Borrelia burgdorferi applied in liposomes and presented to dendritic cells induce CD8+ T-lymphocytes in vitro. Cell Immunol 2000; 201: 124-131.
23. Subair H, Tiwana H, Fielder M, et al. Elevation of anti-Proteus antibodies in patients with rheumatoid arthritis from Bermuda and England. J Rheumatol 1995; 22: 1825-1828.
24. Blankenberg-Sprenkels SHD, Fielder M, Feltkamp TEW et al. Antibodies to Klebsiella pneumoniae in Dutch patients with ankylosing spondylitis and acute anterior uveitis and to Proteus mirabilis in rheumatoid arthritis. J Rheumatol 1998; 25: 743-747.
25. Tiwana H, Wilson C, Walmsley RS, et al. Antibody responses to gut bacteria in ankylosing spondylitis, rheumatoid arthritis, Crohns disease and ulcerative colitis. Rheumatol Int 1997; 17: 11-16.
26. Tani Y, Tiwana H, Kukuda S, et al. Antibodies to Klebsiella, Proteus, and HLA-B27 peptides in Japanese patients with ankylosing spondylitis and rheumatoid arthritis. J Rheumatol 1997; 24: 109-114.
27. Wanchu A, Deodhar SD, Sharma M, et al. Elevated levels of anti-Proteus antibodies in patients with active rheumatoid arthritis. Indian J Med Res 1997; 105: 39-42.
28. Van der Heijden IM, Wilbrink B, Tchetverikov I, et al. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum 2000; 43: 593-598.
29. Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res 2002; 4: Suppl 3S265-72.
30. Kollef MH, West S, Davis DR, et al. Central and peripheral nervous system demyelination after infection with Mycoplasma pneumoniae. Evidence of an autoimmune process. Southern Med J 1991; 84, 1255-1258.
31. Ginsburg KS, Kundsin RB, Walter CW, et al. Ureaplasma urealyticum and Mycoplasma hominis in women with systemic lupus erythematosus. Arthritis Rheum 1992; 35: 429-433.
32. Runge M, Rykena S, Wildhagen K, et al. Detection of Ureaplasma urealyticum in urine of patients with systemic lupus erythematosus and healthy individuals by culture and polymerase chain reaction. J Med Microbiol 1997; 46: 413-418.
33. Komatsu H, Kuroki S, Shimizu Y, et al. Mycoplasma pneumoniae meningo-encephalitis and cerebellitis with antiganglioside antibodies. Pediatric Neurol 1998; 18: 160-164.
34. Yuki, N. Ganglioside mimicry and peripheral nerve disease. Muscle Nerve 2007; 35: 691-711.
35. Kusunoki S, Chiba A, Hitoshi S, et al. Anti-GAL-c antibody in autoimmune neuropathies subsequent to mycoplasma infection. Muscle Nerve 1995; 18: 409-413.
36. Nishimura M, Saida T, Kuroki S, et al. Post-infectious encephalitis with anti-galactocerebroside antibody subsequent to Mycoplasma pneumoniae infection. J Neurol Sci 1996; 140: 91-95.
37. Kuwabara, S. Guillain-Barre syndrome. Current Neurol Neurosci Rep 2007; 7: 57-62.
38. Perera VN, Nachamkin I, Ung H, et al. Molecular mimicry in Campylobacter jejuni: role of the lipo-oligosaccharide core oligosaccharide in inducing anti-ganglioside antibodies. FEMS Immunol Med Microbiol 2007; 50: 27-36.
39. Matyszak MK. Inflammation in the CNS. Balance between immunological privilege and immune response. Progress Neurobiol 1998; 56: 19-35.
40. Speciale L, Sarasella M, Ruzzante S, et al. Endothelin and nitric oxide levels in cerebrospinal fluid of patients with multiple sclerosis. J Neurovirol 2000; 6: S62-S66.
41. Svenningsson A, Petersson AS, Andersen O, et al. Nitric oxide metabolites in CSF of patients with MS are related to clinical course of the disease. Neurology 1999; 53: 1880-1882.
42. Gay, F. Bacterial toxins and multiple sclerosis. J Neurol Sci 2007; 262 :105-112.
43. Gough SCL, Simmonds MJ. The HLA region and autoimmune disease: Associations and mechanisms of action. Current Genomics 2007; 8: 453-465.
44. Parratt J, Tavendale R, O'Riordan, J, et al. Chlamydia pneumoniae-specific serum immune complexes in patients with multiple sclerosis. Multiple Sclerosis 2008; 14: 292-299.
45. Topkaya AE, Sahin S, Aksungar FB, et al. Is there any relationship between streptococcal infection and multiple sclerosis?. Med Sci Monitor 2007; 13: CR567-CR569.
46. Budak F, Keceli S, Efendi H, et al. The investigation of Chlamydophila pneumoniae in patients with multiple sclerosis. Int J Neurosci 2007; 117: 409-415.
47. Casserly G, Barry T, Tourtellotte WW, et al. Absence of Mycoplasma-specific DNA sequence in brain, blood and CSF of patients with multiple sclerosis (MS): A study by PCR and real-time PCR. J Neurol Sci 2007; 253: 48-52.
48. Lindsey JW, Patel S. PCR for bacterial 16S ribosomal DNA in multiple sclerosis cerebrospinal fluid. Multiple Sclerosis 2008; 14: 147-152.
49. Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005; 202: 473-477.
50. Weinshenker BG, Wingerchuk DM, Pittock SJ, et al. NMO-IgG: A specific biomarker for neuromyelitis optica. Dis Markers 2006; 22: 197-206.
51. Bradl M, Lassmann H. Anti-Aquaporin-4 Antibodies in Neuromyelitis Optica: How to Prove their Pathogenetic Relevance? Int MS J 2008; 15:75-78.
52. Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007; 69: 2221-2231.
53. Mycko MA, Cwiklinska H, Walczak A, et al. A heat shock protein gene (Hsp70.1) is critically involved in the generation of the immune response to myelin antigen. Eur J Immunol 2008; 38: 1999-2013.
54. Ebringer A, Wilson C. HLA molecules, bacteria and autoimmunity. J Med Microbiol 2000; 49: 305-311.
55. Beutler AM, Schumacher HR. Reactive arthritis. Is it a useful concept? Br J Clin Practice 1997; 51: 169-172.
56. Lee S, Khare SD, Griffiths MM, et al. HLA-B27 transgenic mice are susceptible to collagen-induced arthritis. Type II collagen as a potential target in human disease. Human Immunol 2000; 61: 140-147.
57. Ringrose JH. HLA-B27 associated spondyloarthropathy, an autoimmune disease based on cross-reactivity between bacterial and HLA-B27? Ann Rheum Dis 1999; 58: 598-610.
58. Zhang HW, Kaur I, Niesel DW, et al. Lipoprotein from Yersinia enterocolitica contains epitopes that cross react with the human thyrotropin receptor. J Immunol 1997; 158: 1976-1983.
59.Luo CY, Seetharamaiah GS, Niesel DW, et al. Purification and characterization of Yersinia enterocolitica envelope protein which induce antibodies that react with human thyrotropin receptor. J Immunol 1994; 152: 2555-2561.
60. Zhang HW, Kaur I, Niesel DW, et al. Yersinia enterocolitica envelope proteins that are corss reactive with thyrotrpin receptor (TSHR) also have B-cell mitogenic activity. J Autoimmunity 1996; 9: 509-516.
61. Hemmerich P, Neu E, Macht M, et al. Correlation between chlamydial infection and autoimmune response. Molecular mimicry between RNA polymerase major sigma subunit from Chlamydia trachomatis and human L7. Eur J Immunol 1998; 28: 3857-3866.
62. Hughes RAC, Hadden RDM, Gregson NA, Pathogenesis of Guillain-Barre syndrome. J Neuroimmunol 1999; 100: 74-97.
63. Rink L, Nicklas W, Luhm J, et al. Induction of a pro-inflammatory cytokine network by Mycoplasma arthritidis-derived superantigen (MAS). J Interferon Cytokine Res 1996; 16: 861-868.
64. Shibata K, Hasebe A, Sasaki T, et al. Mycoplasma salivarum induces interleukin-6 and interleukin-8 in human gingival fibroblasts. FEMS Immunol Med Microbiol 1997; 19: 275-283.
65. Mu HH, Sawitzke AD, Cole BC. Modulation of cytokine profiles by the mycoplasma superantigen Mycoplasma arthritidis mitogen parallels susceptibility to arthritis induced by M. arthritidis. Infection Immunity 2000; 68: 1142-1149.
66. Zurita-Salinas CS, Palacios-Boix A, Yaanez A, et al. Contamination with mycoplasma species induces interleukin-13 expression by human skin fibroblasts in culture. FEMS Immunol Med Microbiol 1996; 15: 123-128.
67. Brenner T, Yamin A, Abramsky O, R. et al. Stimulation of tumour necrosis factor alpha production by mycoplasmas and inhibition by dexamethasone in cultured astrocytes. Brain Res 1993; 608: 273-279.
68. Brenner T, Yamin A, Gallily R. Mycoplasma triggering of nitric oxide production by central nervous system glial cells and its inhibition by glucocorticoids. Brain Res 1994; 641; 51-56.
69. Kikkawa S, Matsumoto M, Sasaki T, et al. Complement activation in Mycoplasma fermentans-induced clearance from infected cells. Probing of the organism with monoclonal antibody against M161Ag. Infection Immunity 2000; 68: 1672-1680.
70. Fabisiak JP, Gao F, Thomson RG, et al. Mycoplasma fermentans and TNF-beta interact to amplify immune-modulating cytokines in human lung fibroblasts. Amer J Physiol-Lung Cell Mol Physiol 2006; 291: L781-L793.
71. Kawahito, Y; Ichinose, S; Sano, H; Tsubouchi, Y; Kohno, M; Yoshikawa, T; Tokunaga D, Hojo T, Harasawa R, et al. Mycoplasma fermentans glycolipid-antigen as a pathogen of rheumatoid arthritis. Bichem Biophys Res Commun 2008; 369: 561-566.
72. Avcin T, Toplak, N. Antiphospholipid antibodies in response to infection. Current Rheumatol Rep 2007; 9:212-218.
73. Moue M, Tohno M, Shimazu T, et al. Toll-like receptor 4 and cytokine expression involved in functional immune response in an originally established porcine intestinal epitheliocyte cell line. Bichem Biochem Acta Gen Subjects 2008; 1780: 134-144.
74. Hasebe A, Mu HH, Washburn LR, et al. et al. Inflammatory lipoproteins purified from a toxigenic and arthritogenic strain of Mycoplasma arthritidis are dependent on Toll-like receptor 2 and CD14. Infection Immunity 2007; 75: 1820-1826.
75. Mu HH, Pennock ND, Humphreys J, et al. Engagement of Toll-like receptors by mycoplasmal superantigen: downregulation of TLR2 by MAM/TLR4 interaction. Cell Microbiol 2005; 7: 789-797.
76. Mu HH, Humphreys J, Chan FV, et al. TLR2 and TLR4 differentially regulate B7-1 resulting in distinct cytokine responses to the mycoplasma superantigen MAM as well as to disease induced by Mycoplasma arthritidis. Cell Microbiol 2006; 8: 414-426.
77. Miggin SM, Palsson-McDermott E, Dunne A, et al. NF-kappa B activation by the Toll-IL-1 receptor domain protein MyD88 adapter-like is regulated by caspase-1. Proc Natl Acad Sci USA. 2007; 104: 3372-3377.
78 Vanhoutte F, Paget C, Breuilh L, et al. Toll-like receptor (TLR)2 and TLR3 synergy and cross-inhibition in murine myeloid dendritic cells. Immunol Lett 2008; 116: 86-94.
80. Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol 2001: 19: 275-90.
81. van Lent PLEM, Blom AB, Grevers L, et al. Toll-like receptor 4 induced Fc gamma R expression potentiates early onset of joint inflammation and cartilage destruction during immune complex arthritis: Toll-like receptor 4 largely regulates Fc gamma R expression by interleukin 10. Ann Rheum Dis 2007; 66: 334-340.
82. Loof TG, Goldmann O, Medina E. Immune recognition of Streptococcus pyogenes by dendritic cells. Infection Immunity 2008; 76: 2785-2792.
83. Okugawa S, Ota, Y, Kitazawa, T, et al. Janus kinase 2 is involved in lipopolysaccharide-induced activation of macrophages. Am. J. Physiol.-Cell Physiol 2003; 285: C399-C408.
84. Marcenaro E, Ferranti B, Falco M, et al. Human NK cells directly recognize Mycobacterium bovis via TLR2 and acquire the ability to kill monocyte-derived DC. Int Immunol 2008; 20: 1155-1167.
85. Tu Z, Bozorgzadeh A, Pierce RH, et al. TLR-dependent cross talk between human Kupffer cells and NK cells. J Exp Med 2008; 205: 233-244.
86. Liese J, Schleicher U, Bogdan C. TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur J Immunol 2007; 37: 3424-3434.
87. Hayem G, De Bandt M, Palazzo E, et al. Anti-heat shock protein 70 kDa and 90 kDa antibodies in serum of patients with rheumatoid arthritis. Ann Rheum Dis 1999; 58: 291-296.
88. Cancino-Diaz ME, Perez-Salazar JE, Dominguez-Lopez L, et al. Antibody response to Klebsiella pneumoniae 60 -kDa protein in familial and sporadic ankylosing spondylitis. Role of HLA-B27 and characterisation as a GroEL-like protein. J Rheumatol 1998; 25: 1756-1764.
89. Al Shamma MRR, McSharry C, McLeod K, et al. Role of heat shock proteins in the pathogenesis of cystic fibrosis arthritis. Thorax 1997; 52: 1056-1059.
90. Musfata AS, Lundin KEA, Meloen RH, et al. HLA-DR4-restricted T cell epitopes from the mycobacterial 60,000 MW heat shock protein (hsp60) do not map to the sequence homology regions with the human hsp60. Immunology 1996; 87: 421-427.
91. Auger I, Escola JM, Gorvel JP, et al. HLA-DR4 and HLA-DR10 motifs that carry susceptibility to rheumatoid arthritis bind to 70-kD heat shock proteins. Nature Med 1996; 2: 306-310.
92. Vinasco J, Beraun Y, Nieto A, et al. Heat shock protein 70 gene polymorphism in rheumatoid arthritis. Tissue Antigens 1997; 50: 71-73.
93. Yokota S, Minota S, Fujii N. Anti-HSP auto-antibodies enhance HSP-induced pro-inflammatory cytokine production in human monocytic cells via Toll-like receptors. Int Immunol 2006; 18: 573-580.
94. Hauet-Broere F, Wieten L, Guichelaar T, et al. Heat shock proteins induce T cell regulation of chronic inflammation. Ann Rheum Dis 2006; 65: 65-68.
95. Binder, RJ, Vatner R.The heat-shock protein receptors: some answers and more questions. Tissue Antigens 2004; 64: 442-451.
96. Calderwood SK, Mambula SS, Gray PJ, et al.. Extracellular heat shock proteins in cell signaling. FEBS Lett 2007: 581: 3689-3694.
97. Singh IS, Gupta A, Nagarsekar A, et al. Heat shock co-activates interleukin-8 transcription. Amer J Resp Cell Mol Biol 2008; 39: 235-242.
98. Brown V, Brown RA, Ozinsky A, et al. Binding specificity of Toll-like receptor cytoplasmic domains. Eur J Immunol 2006; 36:742-753.
99. Luo XJ, Zuo XX, Zhang B, et al. Release of heat shock protein 70 and the effects of extracellular heat shock protein 70 on the production of IL-10 in fibroblast-like synoviocytes. Cell Stress Chaperones 2008; 13: 365-373.
100. Chase MA; Wheeler DS, Lierl KM, et al. Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR4- and NF-kappa B-Dependent mechanism. J Immunol 2007; 179: 6318-6324.
101. Cui XZ, Besch V, Khaibullina A, et al. Neuronal nitric oxide synthase deficiency decreases survival in bacterial peritonitis and sepsis. Intensive Care Med 2007; 33: 1993-2003.
102. Connor JR, Manning PT, Settle SL, et al. Suppression of adjuvant-induced arthritis by selective inhibition of inducible nitric oxide synthase. Eur J Pharmacol 1995; 273: 15-24.
103. Ribeiro-Dias F, Shio MT, Timenetsky J, et al. Mycoplasma arthritidis superantigen (MAM)-induced macrophage nitric oxide release is MHC class II restricted, interferon gamma dependent, and toll-like receptor 4 independent. Exp Cell Res 2003; 286:345-354.
104. Cole BC, Mu HH, Pennock ND, et al. Isolation and partial purification of macrophage- and dendritic cell-activating components from Mycoplasma arthritidis: Association with organism virulence and involvement with toll-like receptor 2. Infection Immunity 2005; 73: 6039-6047.
105. Peltier MR, Freeman AJ, Mu HH, et al. Characterization and partial purification of a macrophage-stimulating factor from Mycoplasma hominis. Amer J Reprod Immunol 2005; 54: 342-351.
106. Lavric M, Bencina D, Kothlow S, et al. Mycoplasma synoviae lipoprotein MSPB, the N-terminal part of V1hA haemagglutinin, induces secretion of nitric oxide, IL-6 and IL-1 beta in chicken macrophages. Vet Mircobiol 2007; 121: 278-287.
107 Hooper DC, Scott GS, Zborek A, et al. Uric acid, a peroxynitrite scavenger, inhibits CNS inflammation, blood-CNS barrier permeability changes, and tissue damage in a mouse model of multiple sclerosis. FESEB J 2000; 14: 691-698.
108. Linares D, Taconis M, Mana P, et al. Neuronal nitric oxide synthase plays a key role in CNS demyelination. J Neurosci 2006; 26: 12672-12681.
109. Kavya R, Saluja R, Singh S, et al. Nitric oxide synthase regulation and diversity: Implications in Parkinson's disease. Nitric Oxide Biol Chem 2006; 15: 280-294.
110. Scarim AL, Heitmeier MR, Corbett JA. Heat shock inhibits cytokine-induced nitric oxide synthase expression by rat and human islets. Endocrinology 1998; 139: 5050-5057.
111. Feinstein DL, Galea E, Aquino DA, et al. (). Heat shock protein 70 suppresses astroglial-inducible nitric oxide synthase expression by decreasing NF kappa B activation. J Biol Chem 1996; 271: 17724-17732.
112. Song MC, Pinsky MR, Kellum JA. Heat shock factor 1 inhibits nuclear factor-kappa B nuclear binding activity during endotoxin tolerance and heat shock. J Crit Care 2008; 23: 406-415.
113. Mangipudy RS, Vishwanatha JK. Role of nitric oxide in the induction of apoptosis by smokeless tobacco extract. Mol Cell Biochem 2000; 200, 51-57.
114. Naito Y, Yoshikawa T, Boku Y, et al. Protective role of intracellular glutathione against nitric oxide-induced necrosis in rat gastric mucosal cells. Alimentary Pharmacol Therapeutics 2000; 14:145-152.
115. Cassidy RA, Burleson DG, Delgado AV, et al... Effects of heme proteins on nitric oxide levels and cell viability in isolated PMNs. A mechanism of toxicity. J Leukocyte Biol 2000; 67, 357-368.
116. Zheng RX, Cohen PA, Paustian CA, et al. Paired toll-like receptor agonists enhance vaccine therapy through induction of interleukin-12. Cancer Res 2008; 68: 4045-4049.
117. Meyer T, Stockfleth E. Clinical investigations of Toll-like receptor agonists. Expert Opinion Invest Drugs 2008; 17: 1051-1065.
118. Himmel ME, Hardenberg G, Piccirillo CA, et al. The role of T-regulatory cells and Toll-like receptors in the pathogenesis of human inflammatory bowel disease. Immunology 2008; 125: 145-153.
119. Owens, RC. Antimicrobial stewardship: concepts and strategies in the 21st century. Diag Microbiol Infectioius Dis 2008; 61: 110-128.
Psychotherapy, psychological treatment and psychological techniques are the motherhood and apple pie of psychiatry. No one can be found to say a bad word against them although the word psychotherapy is often preceded with some qualifier such as “sensible” to indicate that the farther shores of the discipline may not have much place in psychiatry. Sadly though psychotherapy and its congeners, unlike motherhood or even apple pie are is far from widespread in the practice of many psychiatrists and training in the topic is woefully patchy across the country. In this occasional series I hope to introduce the reader to some key concepts in the field not so much from a scholarly perspective in an academic paper decorated with references (although there will be reading for those with sufficient interest and leisure) but from the perspective of practice. I hope to show how each of these psychotherapeutic concepts can be applied both to the practice of formal therapies and to more general aspects of psychiatric practice.
Topic 1 - Alliance
The term alliance refers to the maintenance of a certain kind of positive relationship between the patient and their therapist or doctor. We know that the quality of the alliance in psychotherapy is quite predictive of the likely outcome of treatment so that, while a good alliance does not guarantee a good outcome a bad alliance often ensures a poor outcome. Alliance is something of a portmanteau term since it covers aspects both of trust and liking but also of faith in the skill of the doctor or therapist and a willingness to make positive efforts towards furthering the aims of treatment on the part of the patient. Sometimes this last aspect of alliance – the willingness of the patient to put their best foot forward is referred to by the more descriptive term “working alliance”. Treatments differ in the extent to which they require anything resembling a working alliance. For example many surgical procedures require only that the patient consents and submits to treatment. Other treatments in medicine require that the patient complies with treatment by which is meant carrying out medical instructions accurately. As treatments become more complex and conditions more chronic the degree to which the patient must be an active agent in the administration of their own treatment increases with diabetes being a classic example.
In psychiatry some treatments such as submitting to depot neuroleptic administration require minimal levels of compliance and little in the way of alliance. However other treatments and particularly those which involve making substantial changes in lifestyle require that the patient be almost entirely responsible for the carrying through of their treatment. As such they resemble fitness training or education far more than they resemble “treatments”. In these situations the alliance made between the patient and their doctor is a critical factor in determining the success or failure of treatment.
The making of alliances in ordinary life is not a special skill but something which we all possess however psychotherapists and psychiatrists need to make alliances with people that others shun or who are hostile, ambivalent, distracted or cognitively impaired. The will and the skill to form an alliance with such individuals take training and crucially practice. It can be thought of as comprising three essential parts.
First – preparation.
Second – the interaction (s)
Third – follow up.
Let’s see how these stages play out in a clinical situation. In which a man’s helpers struggle to maintain a fragile alliance.
Rodger was a large and heavily tattooed man. He walked with a rolling gait and with his arms held out from his side as though he was always ready for a fight. He had suffered several head injuries as a younger man and could be both impulsive somewhat unpredictable and volatile. He used drugs and, at bad times would self harm by slashing himself with tin can lids. Staff in the day hospital had managed to engage him to an extent some months ago but he had become enraged when another patient had started winding him up calling him a stupid fathead. He blundered around the unit like an angry bull threw a chair and made threats to kill the other patient. As a result he was excluded from the unit. Rodger simply could not understand why this had happened and asked to see his key worker to make a complaint.
It would certainly be fair to say that the working alliance with Rodger has all but evaporated. The key worker was faced with the task of explaining Rodger’s new situation to him, rebuilding the alliance and possibly defusing an aggressive encounter. She prepared herself in two ways. First she took care with her own safety and the safety of others in the setting. She warned other people she was seeing Rodger and carried a personal alarm. Her objective was to free her mind from too many anxious thoughts about being assaulted as well as to ensure her physical safety. She also prepared herself by reflecting on Rodger’s world imagining it as to him always potentially threatening either physically or psychologically where he felt under threat of being belittled or disrespected in ways he secretly worried but could not afford to admit to himself were true. Last she prepared Rodger by writing to him and telephoning him before the meeting to tell him what was going to happen and why. She made a point of speaking to him in quite formal and respectful terms as “Mr X” and, knowing that he would be very anxious when he arrived she made a point of starting the appointment on time.
The key worker began by asking Rodger what he felt about the meeting and what was on his mind. Starting with the patient’s perspective and seeking to understand things from their point of view is a critical element of forming an alliance. It communicates to the patient that alliance is a two way affair. Rodger began angrily about the whole business and started to wind himself up about the person who had been rude to him and also about the unfairness of being excluded. The key worker agreed that it must feel very unfair to him. Rodger went on crossly that he was always given the “prick tease” invited into places and then chucked out. The key worker agreed that Rodger was often chucked out of things and asked him why he thought that had happened. Rodger said people were down on him and they all picked on him.
In terms of the alliance Rodger and his key worker are already doing better than before. Rodger is able to speak about what is on his mind and the key worker is able to hear it without becoming defensive or frightened. However this is not yet working alliances because the Key worker has not done much other than agree with Rodger’s perspective in as far as it seems correct.
So now the key worker said that he wondered if Rodger had ever thought patients and staff were frightened of him. Rodger bridled and said angrily “there you go you are all the same I don’t care about them what about me. No one asks how I feel.” The key worker had moved too quickly and the alliance, already fragile has collapsed again. So the key worker said. “I have done the same thing as other people do to you, you always feel you get told off and no one ever listens to your point.” Rodger agreed and again warmed to his theme describing the way in which he was always being put down and treated unfairly. As he became more vehement he stood up and began to pace around the room gesticulating. At times he would refer to “them” putting him down but on other occasions he would say “you”. The key worker said, “When you walk around and raise your voice I get frightened of what you might do and it is hard for me to listen to you properly when I feel scared.” Rodger looked startled and sat down abruptly saying rather defensively “I am not going to do anything”
The key worker’s response which was neither threatened nor defensive but tried to state plainly the effect that Rodger’s behaviour was having explicitly referred to the way in which some behaviour can threaten the alliance. The key worker then went on to explain that although Rodger did not feel that his behaviour was threatening other people interpreted it that way. This allowed the key worker to mention the incident in the day hospital again and to say that people had been frightened of Rodger. At this point the Key worker felt Rodger had taken the point and so he suggested they meet again to talk about it some more next week. Notice that the key worker did not try to “close the deal either clinically by, for example making a contract for good behaviour with Rodger or managerially by seeing if Rodger was satisfied by how his complaint had been heard. This was because the key worker judged that these moves might threaten the alliance again and a further outburst could wipe out Rodger’s memory of his new understanding of himself as potentially frightening others and his new understanding of others as frightened.
The key worker followed up on the meeting with Rodger in a number of ways. First by feeding back to the staff at the day hospital, By doing this the key worker was helping them to repair, even in Rodger’s absence, their sense of an alliance with him and maybe preparing the ground for Rodger’s return. The key worker also telephoned Rodger later in the week to find out how he was timing the phone call to a time when Rodger would normally have been in the day hospital. The Key worker began the call by saying “I was thinking about how you might feel today”. By doing this the key worker conveyed to Rodger that Rodger was in his mind even when Rodger himself was not in the room and that Rodger is an object of concern to him. Giving patients the sense of being “held in mind” is crucial to fostering the alliance. When medical staff gives a sense that they do not have the patient in mind there is almost always a severe rupture in the alliance as for example when the doctor starts reading the patient’s notes while they are in the room.
Conclusion
The story of Rodger and his key worker may seem to some over simple. In such natural seeming interactions the skill is cleverly disguised. Although the Key worker appeared spontaneous and appeared not to be considering his words he was in fact weighing them very carefully. He chose language that was appropriate to Rodger’s intellectual level. His statements were brief and contained only a single point. Thus the key worker matched Rodger’s cognitive level. The Key worker also managed the feeling tone in the room very carefully intervening to calm but not truncate potentially explosive feelings and ultimately promoting a little nugget of increased knowledge about the relationships between Rodger and other people. By maintaining an alliance and by carefully moving it into being (even if briefly) a working alliance the key worker managed a little step of progress with Rodger.
COMPETEING INTERESTS
None Declared
AUTHOR DETAILS
CHESS DENMAN, Consultant Psychiatrist in Psychotherapy, Complex Cases Service, Springbank Ward, Cambridge And Peterborough Mental Health Foundation Trust, Fulbourn Hospital, Cambridge, CB15EF
Email: Chess.Denman@cpft.nhs.uk
Follow up Reading:
For those who want a basic text: Oxford Textbook of Psychotherapy, Glen O Gabbard Judith S. Beck & Jeremy Holmes, Oxford University Press 2005
For those who would like more: Cognitive Behaviour Therapy for challenging Problems. Judith Beck, Guildford press New York 2005, Chapters 4,5,6
For the very keen: Safran J. D. & Muran, J. C. (2006). Has the concept of the alliance outlived its usefulness? Psychotherapy, 43, 286-291.
Septic shock still remains one of the leading causes of death in hospital patients. Greater awareness, understanding of the condition .and the knowledge of most effective treatment measures available can decrease the rate of mortality. Making an early, accurate diagnosis of septic shock is the key to increasing survival rates. Excessive inflammation, excessive coagulation and suppression of fibrinolysis are the hallmarks of Sepsis. Infection control, haemodynamic stabilization, and modulation of the septic response are the cornerstones of treatment. The management is influenced more by appropriate treatment with antibiotics and fluids than by specific intensive care. Septic response can be modulated by the use of Steroids and Activated Protein C and with tight glucose control. Low Tidal Volume ventilation and high volume Haemofilteration are other beneficial strategies in Sepsis. As septic shock worsens and fails to respond to all therapy, one must be prepared to limit and withdraw treatment.
Septic shock still remains the one of the leading causes of death in hospital patients. Barely more than 50% of the patients with severe sepsis survive their hospital admission. This unacceptable high mortality can only be reduced if there is greater awareness and understanding of the condition .and the knowledge of most effective treatment measures available. Unplanned admissions to the Intensive Care Unit (ICU) and potentially preventable deaths on wards are associated with a failure to institute early preventive conditions. Greater than 40% of the intensive Care Unit admissions are potentially preventable with improved ward care.
Survival of patients with Septic shock appears to be better if shock develops while the patient is in Intensive Care Unit rather than on general ward despite greater severity of illness in the intensive care group [1].This suggests that the closer observation and earlier treatment can influence the outcome of sepsis.
INCIDENCE:
Septic shock is an increasingly common problem. The incidence of sepsis is increasing year by year. The reasons for this increase are that the people are living longer and this aged population are the most vulnerable to sepsis. We are using advanced technology to sustain life and there has been a rise in the number of immunocompromised patients due to aggressive cancer therapy and the increased prevalence of HIV. The widespread use of broad spectrum antibiotics has increased the rate of both antibiotic resistance and nosocomial infections.
A prospective, multicentre, observational study, recently conducted to evaluate the epidemiology of Sepsis and other characteristics of Intensive Care Unit patients in European countries (called the SOAP study) was endorsed by the European Society of Intensive Care Medicine [2]. This observational study showed a marked difference in the frequency of sepsis between countries, and higher frequencies of sepsis were mirrored by higher mortality rates. (Fig.1)
Fig 1: Incidence of Sepsis in European Countries
There was a direct relationship between the number of organs failing and the Intensive Care Unit mortality. Patients with no organ dysfunction on admission had mortality rates of 6% whereas those with four or more organ failures had mortality rates of 65 %. [2] (Fig. 2)
Fig 2: The SOAP study
As compared to the incidence of other pathologies in Europe the incidence of severe sepsis is higher (32%) [2] (Fig 3)
Fig 3: Incidence of different pathologies in Europe
In septic patients, older age, positive fluid balance, co morbid diseases on admission; cancer and cirrhosis are the most important variables of mortality.
DEFINITIONS
Sepsis is defined as an infection that triggers a particular Systemic Inflammatory Response Syndrome (SIRS). This is characterised by body temperature outside 36oC - 38oC, HR >90 beats/min, respiratory rate >20/min, WBC count >12,000/mm3 or < 4,000/mm3. (Fig 4)
Fig 4: Definitions
There are three recognised stages in the hierarchy of the inflammatory response, with progressively increased risk of organ failure and death. Patients with infections plus two or more elements of the SIRS meet the criteria for sepsis. Those who have end organ failure are considered as having severe sepsis; and those who have refractory hypotension along with the above said criteria are consider to be in septic shock (Fig. 5)
Fig 5: Definitions
PATHOPHYSIOLOGY:
Sepsis is a complex condition starting from an infective stimulus and resulting in an exaggerated immune response. The inflammatory response that was initiated to fight the infection ultimately leads to damage of various organs thorough out the body.
During the onset of sepsis, the inflammatory system becomes hyperactive, involving both cellular and humoral defence mechanisms Endothelial and epithelial cells, as well as neutrophils, macrophages and lymphocytes, produce powerful pro-inflammatory mediators, especially tumour necrosis factor- (TNF-), interleukin (IL)-6, IL-1 and IL-8. Simultaneously, robust production of acute-phase proteins, such as C-reactive protein, occurs and humoral defence mechanisms such as the complement system are activated, resulting in production of pro-inflammatory mediators, including C5a, the complement split product. C5a ultimately enhances cytokine and chemokine production. Furthermore, the coagulation system becomes activated through various mechanisms, often resulting in disseminated intravascular coagulopathy.
The hallmarks of the sepsis are excessive inflammation, excessive coagulation and suppression of fibrinolysis. In addition endogenous Activated Protein C which modulates coagulation, controls inflammation and supports fibrinolysis is also decreased. There is considerable variability in response which is almost certainly to a large degree genetically determined. Those with a tendency to produce excessive cytokines and TNF will have a greater inflammatory response. Simultaneously, the initial vascular damage results in neutrophil activation, neutrophil-endothelial cell adhesion, and further elaboration of inflammatory cytokines. In tissues already prone to dysfunctional oxygen uptake and metabolism, this vascular injury promotes further tissue hypoxia through regional hypo perfusion. This uncontrolled cascade of inflammation and coagulation fuels the progression of sepsis, resulting in tissue hypoxia and ischemia with resultant organ dysfunction and death.
DIAGNOSIS:
Diagnosis of sepsis is not easy. Making an early, accurate diagnosis of septic shock is a key to increasing survival rates. The signs and symptoms of severe sepsis may be subtle. Although the components of SIRS are non specific, the combination of suspected infection and the presence of SIRS may help alert the clinician to a possible diagnosis of sepsis. Although hypotension is another clinical sign that may signal the onset of septic shock, patient may present with sever sepsis and clinically significant global tissue hypoxia in its absence. Metabolic marker such as serum lactate, arterial base deficit may help to identify the severe cases. A single lactate measurement of 4mmol/l or more at initial presentation is associated with an increased rate of mortality [3]. There may well be signs of altered mentation and abnormalities of renal and liver function test, as well as coagulation abnormalities. At least two blood cultures and cultures of other sites as indicated before commencement of antibiotic therapy. Diagnostic studies such as Ultra sound and CT scan should be performed promptly.
D dimmers are grossly elevated in sepsis. Levels of Protein C are lowered which has therapeutic implications. The potential role of biomarkers for diagnosis of infection in patients presenting with severe sepsis remains undefined. Perhaps the most common considerations as diagnostic biomarkers for sepsis have been C-reactive protein and procalcitonin. Despite initial enthusiasm for their potential diagnostic strengths,[4] they have more recently been related to the growing heap of biomarkers that have failed to accurately differentiate sepsis from similar critical illnesses.
The most exciting development in the last 2 years is the recognition of "soluble triggering receptor expressed on myeloid cells-1" (sTREM-1) as a potential biomarker for sepsis. [5] For this marker, a level greater than 60 ng/mL was more accurate than any other clinical and laboratory findings indicating infection
TREATMENT:
The development of new treatment modalities has resulted in a spate of treatment algorithms, often promulgated by medical societies and healthcare improvement organizations. As these modalities have rolled out, increasing levels of evidence have emerged to support or refute their utility in treating patients with sepsis. One of the greatest endeavours to date is the Surviving Sepsis Campaign (SSC) [6] that was originally launched in 2002 with the stated goal to reduce mortality by 25%. The primary method to achieve this goal was the development of evidence-based sepsis care guidelines that were published in 2004. [6] and recently revised in 2008.
The Institute for Healthcare Improvement (IHI) has highlighted sepsis as an area of focus and has identified several deficiencies that may cause suboptimal care of patients with severe sepsis. These deficiencies include inconsistency in the early diagnosis of severe sepsis and septic shock, frequent inadequate volume resuscitation without defined endpoints, late or inadequate use of antibiotics, frequent failure to support the cardiac output when depressed, frequent failure to control hyperglycemias adequately, frequent failure to use low tidal volumes and pressures in acute lung injury, and frequent failure to treat adrenal inadequacy in refractory shock.
The management of patient with sepsis is influenced more by appropriate treatment with antibiotics and fluids than by specific intensive care. Therefore early intervention should never be delayed pending admission to the intensive care unit. The early and aggressive treatment of septic shock has been well documented in the survival sepsis campaign which is based on the best current practice.
The cornerstones of treatment are infection control, haemodynamic stabilization, and modulation of the septic response.
1. Infection Control:
Infection control is vital if the patient is to have any chance of survival. Appropriate broad-spectrum antibiotics must be given within the first hour of recognition of sepsis after obtaining various cultures. Evidence clearly shows that delay or inadequate antibiotic treatment results in poorer outcome. For every hour lost mortality climbs by 9%. [7]
Initial empirical anti-infective therapy should include one or more drugs that have activity against all likely pathogens (bacterial and/or fungal) and that penetrate in adequate concentrations into the presumed source of sepsis[8] antimicrobial regimen be reassessed daily to optimize activity, to prevent the development of resistance, to reduce toxicity, and to reduce costs
A focus of infection must be sought for and if discovered dealt with immediately. The patient should be evaluated for a focused infection amenable to source control measures including abscess drainage or tissue debridement. One must weigh up the benefits and risks of the particular procedure chosen. If intravascular devices are a potential source, they must be promptly removed after establishing other vascular access. When source control is required, the effective intervention associated with the least physiologic insult be employed (e.g., percutaneous rather than surgical drainage of an abscess)
2. Haemodynamic Stabilization:
In septic shock there is extensive cardiovascular derangement. Hypotension is caused by myocardial depression, pathological vasodilatation and extravasation of circulating volume due to widespread capillary leak. The initial resuscitative effort is to attempt to correct the absolute and relative hypovolemia by refilling the vascular tree. There is no evidence to support one type of fluid crystalloid or colloid is superior to the other. There is good evidence that early gold directed aggressive volume resuscitation improves outcome of sepsis[9] During the first 6 hours of resuscitation the goals of initial resuscitation are a Central venous pressure of 8-12 mm Hg, Mean arterial pressure (MAP) ≥ 65 mmHg, Urine output ≥ 0.5 mL • kg-1 • hr and a central venous (superior vena cava) or mixed venous oxygen saturation ≥ 70% or ≥ 65%, respectively The Rivers study clearly shows a reduction in hospital mortality, 28 day mortality as well as 60 day mortality attributed to the Early Goal Directed Therapy (EGDT) [10]. Early goal-directed resuscitation has been shown to improve survival for emergency department patients presenting with septic shock in a randomized, controlled, single-centre study.[11] Resuscitation directed toward the previously mentioned goals for the initial 6-hr period of the resuscitation was able to reduce in hospital, 28-days as well as 60 days mortality rate (Fig. 6).
Fig 6: Results of Early Goal Directed Therapy (EGDT)
If Scvo2 or SVo2 of 70% or 65%, respectively, is not achieved with fluid resuscitation to the central venous pressure target, then transfusion of packed red blood cells to achieve a hematocrit of ≥ 30% and/or administration of a dobutamine infusion (up to a maximum of 20 µg • kg-1 • min-1) be used to achieve this goal.
It is important to remember that vasopressors should be utilized not only when fluids fail to reverse hypotension, but also during resuscitation to maintain minimally adequate blood pressure. Traditionally, the use of noradrenalin in patients with shock has been restricted by the fear of excessive vasoconstriction that may result in end-organ hypo perfusion. In the past it was usually given only when other vasopressin agents failed, and thus such patients would be predicted to have a poor outcome. Recent studies indicate that the fear of deleterious effect was unwarranted and that noradrenalin may have a role as a first-line vasopressor agent in patients with septic shock.
Vasopressin should be considered in refractory shock despite high dose conventional vasopressors. Vasopressin is an endogenously produced hormone that is deficient in many patients with septic shock. Exogenously administered vasopressin in physiologic replacement doses may act synergistically with other vasopressor agents, and has been associated with early withdrawal of catecholamine. Most studies have evaluated short-term infusions of vasopressin at 0.08 U/minute or less as add-on therapy in patients requiring adrenergic agents. The results show that starting vasopressin in patients with septic shock increases systemic vascular resistance and arterial blood pressure, thus reducing the dosage requirements of adrenergic agents [12]. These effects are rapid and sustained. Substantial enhancement of urine production, likely due to increased glomerular filtration rate, was shown in several studies. A few studies demonstrated clinically significant reduced cardiac output or cardiac index after vasopressin was begun, necessitating cautious use in patients with cardiac dysfunction.
3. Modulation of Septic Response:
There are a number of ways to modulate the septic response. These includes use of steroids, tight glucose control and the use of Activated Protein C. Septic shock causes adrenal suppression and this can be confirmed by measuring cortisol levels or by using the synacthan test. Compare to placebo, the administration of low dose of hydrocortisone (200-300 mg/day in divided doses) to patients with septic shock decrease there requirements for vasopressors [13] and lowered their mortality rate [14]. Low dose hydrocortisone should only be given to non responders of the synacthan test but in practice all patient receive this treatment until the result of the test are received. Following the Corticosteroid Therapy of Septic Shock (CORTICUS) study there is now an increasing trend towards restricting the use of low dose hydrocortisone only to patients with refractory hypotension who are already on high doses on vasopressors [15]. The trial did show a faster resolution of septic shock in patients who received steroids but failed to show a mortality benefit with steroids therapy. Close control of blood glucose has been shown to increase survival in critically ill septic patient. When conservative (10 – 11.1 mol/L) glycemic control was compared with tight control (4.4-6.1mmol/L) in a multi centre, randomized controlled trial, tight control lead to a significant reduction in mortality (8% versus 4-6%), p < 0-04 and improved morbidity at 12 months [16].
Activated Protein C
Human activated Protein C (APC) is an endogenous regulator of coagulation. In order for protein C in the plasma to become activated, it must combine with thrombin and thrombomodulin along with the endothelial protein C receptor. With endothelial damage this activation does not take place resulting in its deficiency. Therefore APC supplementation is a rational therapeutic option. APC has an important role in the management of severe sepsis. It protects against the disruption of the endothelial cell membrane, improves micro circulatory perfusion, and has anti inflammatory, procoagulant, fibrinolytic and anti apoptotic activity. APC must ideally be started with in the first 24 hours of the onset of septic shock. The Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis (PROWESS) trial found activated protein C to reduce the risk of death among all severe sepsis patients by 20% [17] This study has also recognized the risk of complications specially haemorrhages.
Subsequent studies have shown similar results. “Administration of Drotrecogin Alfa (Activated) in Early Stage Severe Sepsis” (ADDRESS trial) also provides the evidence concerning use of rhAPC in adults [18]. Additional safety information comes from an open-label observational study, “Extended Evaluation of Recombinant Activated Protein C” (ENHANCE trail). [19] The ENHANCE trial also suggested that early administration of rhAPC was associated with better outcomes.
Other beneficial Strategies in Sepsis
Low Tidal Volume Ventilation: using normal or high tidal volume (10-12mls/Kg) ventilation will cause over expansion of the normal lung segments. This will in turn result in inflammatory mediators being released in the lung tissue. The consequences of this are the development of Acute Lung Injury (ALI) or Acute Respiratory Distress Syndrome (ARDS). Therefore it is crucial to use low tidal volume Ventilation (6ml/kg) to keep plateau airway pressure less than 30 cm of water [20, 21]
High volume Haemofilteration: In the past five years, many studies have been conducted to evaluate and demonstrate benefits of increasing the volume of ultra filtration and replacement fluid during Continuous Renal replacement therapy [22, 23] particularly in complex and very severe syndromes such as Severe Sepsis and Septic Shock, associated with or without acute renal failure.
In general, the high-volume approach provides higher clearances for middle/high molecular weight solutes than a simple diffusive transport, Continuous veno venous haemodialysis (CVVHD) or a convection-based transport at lower volumes, Continuous veno venous haemofiltration (CVVH). These solutes seem to be primarily involved in the Systemic Inflammatory Response Syndrome, which characterizes the Sepsis syndrome, and their efficient removal may thus be beneficial. [24]
Alternative approaches have been based on more efficient removal of inflammatory mediators by high cut-off hemofilters, which are characterized by an increased effective pore size. Most commercially available hemofilters do not permit a substantial elimination of cytokines because of the low cut-off point of their membranes. The use of high cut-off hemofilters is a new and effective approach to cytokine removal, but it has potentially harmful side effects, such as the loss of essential proteins like albumin [25].
Because the reversibility of this disease and the resultant mortality may be greatest during the earliest stages of presentation, proper sepsis management should not be confined within the walls of an Intensive Care Unit. Specific emphasis on appropriate triage to ensure prompt diagnosis of the high-risk patient is vital to the launch of a coordinated and cooperative effort by the primary treating clinician and the intensivist
Ethical Dilemmas in Septic Shock
Patient with septic shock have a high mortality and as yet there is no predictive scoring system which gives accurate predictions of outcome for individual patient. Survival from an episode and septic shock is dependent on patient’s age, number of failed organs, previous health and the time delay before the onset of medial intervention, as well as the appropriateness and quality of medical care. The resources available to us are not limitless and so difficult decisions have to be made deciding between the potential benefits for one critically ill patient and need for several less critically ill patients. As an intensivist one must set realistic expectations which must be clearly communicated to the families concerned. As septic shock worsens and fails to respond to all therapy, one must be prepared to limit and withdraw treatment.
COMPETING INTERESTS
None Declared
AUTHOR DETAILS
KHADIJA E QURESHI, BSC, MBBS, DA, FCPS, ST2- Anaesthetics, Hemel Hempstead General Hospital, United Kingdom
ABID RAJAH, MB ChB, FRCA, FFARCSI, Lead Clinician & Consultant in Intensive Care, Hemel Hempstead General Hospital, United kingdom
CORRESPONDENCE: Dr Abid Rajah, Lead Clinician & Consultant in Intensive Care, Hemel Hempstead General Hospital, Hill field Road, Hemel Hempstead, HP2 4AD
Email: ARajah@aol.com
REFERENCES
Lundberg J, Perl TM. Septic shock: an analysis of outcomes for patients with onset on hospital wards vs. Intensive care units. Crit care Med 1998; 26: 1020-24
Vincent JL, Sakr Y, and Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006; 34:344-353.
Shapiro NI, Homel MD, Talmor D, Nathanson LA, Lisbon A, Wolfe RE, et al. Serum lactates as a predictor of mortality in emergency department patient with infection. Ann Emerg Med. 2005; 45: 524-8.
Harbarth S, Garbino J, Pugin J, et al. Inappropriate initial antimicrobial therapy and its effect on survival in a clinical trial of immunomodulating therapy for severe sepsis. Am J Med. 2003; 115:529-535.
Gibot S, Kolopp-Sarda MN, Bene MC, et al. Plasma level of a triggering receptor expressed on myeloid cells-1: its diagnostic accuracy in patients with suspected sepsis. Ann Intern Med. 2004; 141:9-15.
Surviving Sepsis Campaign. Available at: http:// www.surviving sepsis.org/ Accessed March 6, 2007.
Dellinger RP, Carlet JM, Masur H, et al. Surviving Sepsis C guidelines for management of severe sepsis and septic shock. Crit Care med. 2004;32: 858-873
Battleman DS, Callahan M, Thaler HT. Rapid antibiotic delivery and appropriate antibiotic selection reduce length of hospital stay of patients with community acquired pneumonia: link between quality of care and resource utilization. Arch Intern Medicine 2002; 162(6):682-8.
Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003; 31:1250-1256.
Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368-1377
Emanuel Rivers. The outcome of patients presenting to the emergency department with severe sepsis or septic shock. Crit Care. 2006; 10(4): 154.
Obritsch MD; Bestul DJ; Jung R; Fish DN; MacLaren R. The role of vasopressin in vasodilatory septic shock. Pharmacotherapy. 2004; 24(8):1050-63 (ISSN: 0277-0008)
Briegel J, Frost H, Haller M, Shelling G, Kilger E, Kuprat G et. Stress doses of hydrocortisone reverse hyper dynamic septic shock: a prospective randomised double blind single centre study. Crit care Med 1999; 27:723-32.
Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002; 288:862-871.
Sprung CL. Update on clinical trials in severe sepsis. CORTICUS trial. Program and abstracts of the Society of Critical Care department patient with infection. Ann Emerg Med 2005: 45 (524-8).
Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449-461
Bernard GR, Vincent JL, Laterre PF, et al. Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001; 344:699-709.
Abraham E, Laterre PF, Garg R, et al: Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med 2005; 353:1332-1341.
Vincent JL, Bernard GR, Beale R, et al: Drotrecogin alfa (activated) treatment in severe sepsis from the global open-label trial ENHANCE: Further evidence for survival and safety and implications for early treatment. Crit Care Med 2005; 33:2266-2277.
ARDS Network Investigators. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000; 342:1301-1308.
National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, Wiedemann HP, Wheeler AP, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006; 354:2564-2575.
Piccinni P, Dan M, Barbacini S, Carraro R, Lieta E, Marafon S, Zamperetti N, Brendolan A, D'Intini V, Tetta C, et al.: Early iso-volaemic haemofiltration in oliguric patients with septic shock. Intensive Care Med 2006, 32:80-86.
Reiter K, D'Intini V, Bordoni V, Baldwin I, Bellomo R, Tetta C, Brendolan A, Ronco C: High-volume hemofiltration in sepsis. Nephron 2002, 92:251-258.
Ronco C, Tetta C, Mariano F, Wratten ML, Bonello M, Bordoni V, Cardona X, Inguaggiato P, Pilotto L, d'Intini V, Bellomo R: Interpreting the mechanisms of continuous renal replacement therapy in sepsis: the peak concentration hypothesis. Artif Organs 2003, 27:792-801.
Abbreviations BSCCP British Society of Colposcopy and Cervical Pathology
CIN Cervical intraepithelial neoplasia
CGIN Cervical glandular intraepithelial neolpasia
DNA Deoxyribonucleic acid
HPV Human papillomavirus
HSIL High-grade squamous intraepithelial lesion
LBC Liquid-based cytology
LLETZ Large loop excision of transformation zone
LSIL Low-grade squamous intraepithelial lesion
NHSCSP National Health Service Cervical Screening Programme
Introduction
Papanicolaou’s publication in 1940s, which showed that exfoliated cervical cells could be reliably harvested and spread, fixed and stained on a glass slide, laid the foundations of cervical screening.
In the last two decades, there has been immense progress in the understanding of cervical carcinogenesis and the currently accepted view is that HPV is an essential factor in the causation of the disease. If HPV is persistent, integration into the cellular genome may occur, which results in the inactivation of tumour suppresser genes, suppression of apotosis, genetic instability and development of precancerous change. Additional genotoxic agents, such as smoking, contribute further to the progression of cervical cancer.
The death rate from cervical cancer was essentially unchanged until the national programme was instituted in 1988. The White Paper The Health of the Nation set a national target to reduce the mortality from cervical cancer by at least 20% by the year 2000 (from 15 per 100,000 populations in 1986 to no more than 12 per 100,000, directly standardize against the European population). The NHS Cervical Screening Programme (NHSCSP) exceeded the target by the year 1997, when the rate fell to 8.9 per 100,000. It continues to fall.
Cervical screening programme1
The programme originally involved every woman between the ages of 20 and 64 years (20-60 years in Scotland) being called and recalled every 3-5 years for a cervical smear test. The evidence has indicated that a more effective screening programme can be offered to women by changing the frequency of screening according to a woman’s age. In 2004, the NHSCSP has issued guideline number 20 which covers all of the major aspects of screening, diagnosis, treatment and follow up.
Age group (years)
Frequency of screening
25
First invitation
25 – 49
Three yearly
50 – 64
Five yearly
65+
Only screen those who have not been screened since age 50 or those who have had recent abnormal tests
Age at starting screening
The incidence of cervical cancer under the age of 25 years is low, and the prevalence of transient HPV infection is high. Much of this prevalent disease would resolve spontaneously. Hence, screening women under the age of 25 years may do more harm than good (unnecessary attendance to colposcopy clinic, increased anxiety and possible over treatment).
Screening interval
A 2003 publication indicated that, to be effective in younger women, screening needs to be more frequent. Therefore, the new screening intervals are to be 3 yearly until the age of 50 years when 5 yearly screening until the age of 64 years, because the most incidences of CIN will have been prevented by prior screening2.
Age at finishing screening
The prevalence of CIN3 and invasive cancer in women over the age of 50 is low. Although it is possible that it may be safe to withdraw well screened women with a negative smear history from screening programme at age 50 years, there is no robust evidence to withdraw this level of healthcare.
Population coverage
A major success in the cervical screening programme has been to increase population coverage. There remain certain women who do not participate, including some ethnic minorities and some women who choose not to. A significant proportion of women who develop cancer have not been regularly screened. Additional effort is required to convince some women that screening can be life saving.
LBC
Liquid base cytology provides almost total elimination of inadequate smear. The UK pilot studies concluded that inadequate cytology would be cut by 87 %, from 9.1% with Pap slides to an average of 1.6 % with LBC.
It has been established from systematic reviews that routine primary cervical screening carries a 50 – 70 % sensitivity to detect CIN3. LBC increases overall sensitivity, gives rise to less equivocation in low grade smear and leads to less referral for colposcopy. There is no difference between the specificity of LBC and Pap smear3.
Smear reports
Acceptable range4
Negative smear
Number of abnormal smear
8.1 – 8.3%
Inadequate smears
5.8 – 12.9%
Borderline nuclear abnormality Mild Dyskaryosis
4.1 – 9.5%
Moderate & Severe dyskaryosis
1 – 2 %
Referral guideline for colposcopy
Women with the following smear results should have the colposcopy assessment.
3 consecutive inadequate smears
3 borderline changes in squamous cells
3 abnormal smears at any grade in a 10 year period
1 borderline change in endocervical cells
1 or 2 mild dyskaryosis (1 mild change – acceptable to repeat a smear)
1 moderate dyskaryosis
1 severe dyskaryosis
1 abnormal glandular smear
Time interval: referral – colposcopy
Abnormal smear
within 8 weeks
Moderate or severe dyskaryosis
within 4 weeks
Glandular abnormality or possible invasion
within 2 weeks
Treatment
Recent evidence suggests that possibly all major-grade (CIN 2, CIN 3, HSIL) lesions should be treated, whereas minor-grade (CIN 1, LSIL) lesions should be managed more conservatively.
Over the last decade the trend has been directed toward more conservative methods of managing CIN. This has coincided with the introduction of the large loop diathermy excision technique. A large multicenter study covering over 13000 treatments has recorded the continuing small risk of patients treated with conservative modalities to develop invasive cancer many years after initial treatment. The risk was still present up to 14 years following treatment.
Method of treatment
Local destructive techniques
It is imperative that any such method destroys the CIN contained within the cervical glands or, more correctly the crypts. Therefore, to be totally effective, these methods must destroy the tissue to the depth of at least 6-7 cm. These methods are the treatment of choice for selected cases in which the entire abnormality is visible on the ectocervix, and in which there is no suggestion of invasion. The principal disadvantage of this method is that a histologic examination of the entire lesion is not possible and early invasive cancer may remain undetected. There are four local destructive techniques:
Cryotherapy, or freezing the area by the application of probes; anaesthesia is not usually required.
Cold coagulation, usually without, or with some local anaesthesia.
Electrodiathermy, under either local or general anaesthesia.
Carbon dioxide (CO2) laser evaporation, usually with local analgesia.
Excisional techniques
Cold knife biopsy
Laser cone biopsy
Large loop diathermy
Hysterectomy: abdominal or vaginal
The optimal method of CIN treatment
There is no obviously superior conservative surgical technique for the treatment of CIN. Excisional treatments permit histological assessment of biopsy and can determine risk factors for residual disease.
The studies have led many authors to advocate the use of excision rather than local destruction techniques as the loops have discovered early invasive lesion in excised specimens. But it can be argued that many of the early micro-invasive lesions now found by the use of excision techniques would have been quite effectively destroyed by the use of local destructive techniques. Now at the moment, it must be left to the individual clinician to choose which technique gives the best results.
Complication
Immediate
The morbidity for excisional method is 2-4 % with immediate discomfort and bleeding.
Long term
Cervical stenosis and constriction: This problem tends to occur most frequently in postmenopausal and post partum women, and result in the development of pyometra. In the younger woman, the stenosis may lead to pelvic endometriosis following on haematometra. The patient often presents with symptoms of painful and prolonged menstruation. The simple management is to perform a dilatation of cervix under general anaesthesia. Even use of a narrow endocervical brush may relieve the symptom.
Excessive eversion of the columnar epithelium : It is not uncommon for the cervix to appear with a large area of exposed columnar epithelium, especially after cone biopsy. Such a situation may result in complaints of postcoital and intermenstrual bleeding or discharge. Nevertheless, it is possible for this exposed transformation zone to become infected yet again with mutagenic agent that resulted in the development of CIN.
It may be necessary to stimulate metaplasia of this area by applying cryosurgery, cautery, or even laser vaporization to columnar epithelium. However, for most patients active treatment is not necessary.
Subsequent pregnancy: There is always concern about subsequent fertility and pregnancy outcome following treatment for CIN. The morbidity associated with the excision of a small fully visible TZ will be different from that associated with a large zone which extends 2 cm up the endocervical canal.
The evidence found no effect on subsequent fertility and pregnancy outcome following loop diathermy treatment. However, it is found to have a higher incidence of low birth-weight babies when compared with controls. More recently, other authors have shown that using the CO2 laser, a cone biopsy greater than 10 mm in depth acts as an independent risk factor for the occurrence of preterm labour5.
Success rate
Modern conservative therapies for the treatment of CIN are extremely successful, with the clearance rate in the order of 95 % or better, except cryosurgery which has a lower clearance rate than other conservative method (85%).
Recurrence
The rate of dyskaryosis in 12 months following both LLETZ and laser ablation was 4.4 %. A cumulative rate of recurrence at 4 years was 10.1 per 100 women.
Follow up
Follow up after conservative treatment
Women aged 50 years or more with positive excision margin are particularly at risk of persistent and recurrent disease. Cytology alone is recommended for follow up and should start at six month following treatment.
Women treated for high grade disease (CIN2, CIN3, CGIN) require 6 and 12 month follow up cytology and annual cytology for subsequent nine years before returning to screening at routine interval.
Women treated for low grade disease require 6, 12 and 24 month follow up cytology. If all results are negative, then women may return to screening at routine interval.
Women treated for CGIN are at higher risk of developing recurrent disease than those with high grade CIN. Ideally, six-monthly samples would be taken for five years followed by annual samples for a further five years.
Follow-up after hysterectomy
Women who have had a hysterectomy with CIN present are potentially at risk of developing vaginal intraepithelial neoplasia (incidence 1%) and invasive vaginal disease.
For women on routine recall for at least 10 years prior to hysterectomy and no CIN in the sample at hysterectomy, no vault cytology is required.
For women with less than 10 years’ routine recall and no CIN at hysterectomy, a sample should be taken from the vault six months after surgery and there should be no further cytology follow-up if it is negative.
For women with completely excised CIN at hysterectomy, a sample should be taken from the vault at 6 and 18 months after surgery and there should be no further cytology follow-up if both are negative.
For women with incomplete or uncertain excision of CIN, follow-up should be conducted as if the cervix is still in situ.
Summary of follow up
Histology/ Pre-treatment smear history
Follow up
After conservative treatment
Low grade CIN
6, 12 and 24 months and then routine screening
After conservative treatment
High grade lesion (CIN2, CIN3, CGIN)
6, 12 and annual cytology for 9 years and then routine screening
After hysterectomy
Routine recall in last10 years, No CIN
No vault smear
After hysterectomy
Less than 10 years, Routine recall, No CIN
Vault smear 6 months after hysterectomy
After hysterectomy for CIN
Complete excision of CIN
Vault smear 6 and 8 months after hysterectomy
After hysterectomy for CIN
Incomplete or uncertain excision of CIN
Follow up as if the cervix is still in situ
The potential role of HPV testing
The type II hybrid capture is a new method for the detection of HPV DNA in cervical mucosa. The following list of clinical uses of hybrid capture is suggested:
As a screening method, together with cytology:
For patients with abnormal cytology, to select patients who will be referred to a colposcopic clinic.
To evaluate the low-grade lesions forecast.
The use of hybrid capture as a screening method is based on the principle that the cytology has a sensitivity of approximately 56%, and the sensitivity of virus typification is 77%; but using both at the same time, the diagnostic sensitivity amount to 93%. Whether hybrid capture should be used as a screening method is still being debate6. A recent RCT7 reported that adjunctive HPV testing did not add significantly to the effectiveness or cost effectiveness of LBC to the detection of CIN 3.
Vaccination against cervical cancer8
Without further preventive measures, death from cervical cancer are predicted to jump four-fold to over a million a year by 2050 as a result of the explosion in HPV infection rates across the world. Vaccination as a primary prevention has obvious advantages in countries where screening programmes are not established but may also offer advantages in countries like the UK, where secondary prevention by screening and treating premalignant lesions is not only expensive but sometimes imprecise, resulting in unnecessary anxiety and intervention for some women, while at the same time failing to detect lesions in others.
Rationale
Women previously infected with a particular HPV type are unlikely to become reinfected by the same type, because of immunity largely provided by antibodies targeted against the major papillomavirus capsid protein L1. When made in the laboratory, L1 protein self-assembles into virus-like particles (VLPs) that are morphologically identical to HPV and highly immunogenic but not in themselves infectious because of lack of viral genome.
Gardasil (Merk) is a quadrivalent vaccine offering protection against HPV types 6, 11, 16 and 18. The longevity of this immune response varied, with only 76% of vaccines showing detectable antibody response to 36 months after immunisation. There is preliminary evidence of cross protection against infection with related HPV 31 and 45. Gardasil and Cervarix has an excellent safety record with only transient injection site reaction and no evidence of adverse effects on chronic disorders.
In the UK, the HPV vaccination programme targets the girls from 12 to 13 year old and additional programme for the girls from 13 to 18 years old, starting in September 2008 and finishing in 2011. HPV-specific antibodies generated by vaccination may wane with time, although current data indicate that immune responses persist through 5 years. The need for booster immunisations to maintain protection against infection will become apparent after prolonged periods of follow up.
The abnormal smear in pregnancy
Ten to fifteen in 1000 pregnant women have their smear abnormal. Recommendations for referral colposcopy are the same in pregnancy as in non-pregnant women. Much more reassurance is required, with emphasis on the fact that the colposcopy will not harm the fetus or cause miscarriage. The treatment for preinvasive lesions may be postponed until after delivery. The essential role of biopsy is to rule out an invasive disease.
The cervical smear in menopausal women
Oestrogen deficiency causes atrophy of tissue and a retraction of squamocolumnar junction. The epithelium becomes thinner and more easily traumatized. There is a greater incidence of unsatisfactory smear reports and unsatisfactory colposcopy. It is generally preferable to repeat smear after oral, transdermal or vaginal estradiol for a period of 7 to 10 days.
Conclusion
The cervical smear is a simple and effective screening which has a number of deficiencies. False-negative smears are principally due to imperfect sampling, errors of cytological interpretation, and in rare cases to rapid progression of lesions in sites which are difficult to access. New technologies can improve the sensitivity of screening. The emphasis is on developing systems that will screen for preinvasive stage of cervical cancer and thereby allow assessment and appropriate management.
COMPETING INTERESTS
None Declared
AUTHOR DETAILS
TINT TINT WAI, ST3, Obstetrics and Gynaecology Department, Bedford Hospital, United Kingdom
DILIP PATIL, Consultant Obstetrician and Gynaecologist, Bedford Hospital, United Kingdom
CORRESPONDENCE: D Patil, Consultant Obstetrician and Gynaecologist, Bedford Hospital, United Kingdom,
Email: patild@yahoo.com
References
Colposcopy and Programme Management. NHS Cervical Screening Programme publication 20. April 2004
Progress in cervical screening. Scientific Advisory Committee. RCOG Opinion paper 7. June 2006
Guidance on the use of LBC for cervical screening. NICE, Technology appraisal 69. October 2003
David M Luesley, Mohamood I. Shafi and Joseph A. Jordan. Handbook of Colposcopy; Second edition 2002
Albert Singer & John M Monaghan. Lower Genital Tract Precancer. Second edition 2000
W Prendiville, J Ritter, S Tatti, L Twiggs. Colposcopy management options. First edition 2003
Henry Kitchener, et al. ARTISTIC: A randomised trial of HPV testing in primary cervical screening: Final results; BSCCP Annual Scientific Meeting, book of abstracts, 2008
Vaccination against cervical cancer, Scientific Advisory Committee, Opinion paper 9, RCOG; February 2007
We are in the middle of the greatest changes in mental health law in a century. This started with the introduction of the Mental Capacity Act in October 2007, continued with amendments to the Mental Health Act (MHA) in November 2008, and will finish with a final flourish when the Deprivation of Liberty Safeguards come into effect to April 2009.
But what do these changes mean for Psychiatrists and other doctors? This article explores the changes.
The Mental Health Act 1983 (as amended by the Mental Health Act 2007)
Although the structure of the Mental Health Act remains the same – for example, compulsory admission to hospital still requires the recommendations of 2 doctors and the agreement of an Approved Mental Health Professional (the replacement of the ASW), the Act has been amended in the following ways:
Key Change 1
Introducing a Simplified Single Definition of Mental Disorder.
Key Change 2
Abolishing the ‘Treatability’ Test and introducing a new Appropriate Medical Treatment Test.
Key Change 3
Ensuring that Age Appropriate Services are available to any patients admitted to hospital who are aged under 18 (anticipated by 2010).
Key Change 4
Broadening the Professional Groups that can take particular roles.
Key Change 5
Introducing the right for patients to apply to court to displace their Nearest Relative, and including civil partners in the list of potential nearest relatives.
Key Change 6
Ensuring that patients have a right to an Advocacy Service when under compulsion (implemented in 2009).
Key Change 7
Introducing new safeguards regarding Patients and Electro-Convulsive Therapy.
Key Change 8
Introducing a new provision to allow Supervised Community Treatment. This allows a patient detained on a treatment order to receive their treatment in the community rather than as an in-patient.
Key Change 9
Earlier automatic referral to a Mental Health Review Tribunal (Tribunal) where patients don’t apply themselves. & new Tribunal system structure.
Key Change10
New ‘2nd Professional’ role for renewal of section 3.
All these changes need to be seen within the context of the Guiding Principles and the clarified legal status of the Code of Practice (COP).
The MHA tells us WHAT to do
The COP explains HOW to do it
The Guiding Principles help us to apply the MHA and COP in INDIVIDUAL SITUATIONS
The status of the Code of Practice
The Act makes it clear that professionals, including doctors, are expected to follow the guidance of the Code of Practice, or explain why they haven’t done so. It also makes it clear that professionals are expected to take account of a number of issues, the guiding principles, when making decisions.
The Principles:
Purpose
Decisions under the Act must be taken with a view to minimising the undesirable effects of mental disorder, by maximising the safety and well-being (mental and physical) of patients, promoting their recovery and protecting other people from harm.
Least restriction
People taking action without a patient’s consent must attempt to keep to a minimum the restrictions they impose on the patient’s liberty, having regard to the purpose for which the restrictions are imposed.
Respect
People taking decisions under the Act must recognise and respect the diverse needs, values and circumstances of each patient, including their race, religion, culture, gender, age, sexual orientation and any disability. They must consider the patient’s views, wishes and feelings (whether expressed at the time or in advance), so far as they are reasonably ascertainable, and follow those wishes wherever practicable and consistent with the purpose of the decision. There must be no unlawful discrimination.
Participation
Patients must be given the opportunity to be involved, as far as is practicable in the circumstances, in planning, developing and reviewing their own treatment and care to help ensure that it is delivered in a way that is as appropriate and effective for them as possible. The involvement of carers, family members and other people who have an interest in the patient’s welfare should be encouraged (unless there are particular reasons to the contrary) and their views taken seriously.
Effectiveness, efficiency and equity
People taking decisions under the Act must seek to use the resources available to them and to patients in the most effective, efficient and equitable way, to meet the needs of patients and achieve the purpose for which the decision was taken.
In the future, it is likely that doctors will be expected to justify decision making with reference to above principles.
Key Change 1: Definition of Mental Disorder:
The definition of disorders covered by the MHA is now “any disorder or disability of the mind”. This simplified definition now applies to all sections of the Act. The four forms of mental disorder (mental illness, mental impairment, severe mental impairment and psychopathic disorder) have disappeared. This potentially means some people previously excluded are now included. For example, there may be some people with an acquired brain injury who were not covered by the term “mental impairment or severe mental impairment” who could now benefit from the protections of the Act.
The Learning Disability Qualification has been introduced to preserve the status quo (e.g. under section 3, a person with a learning disability alone can only be detained for treatment or be made subject to Guardianship if that learning disability is associated with abnormally aggressive or seriously irresponsible conduct.) and now applies to all those sections that relate to longer-term compulsory treatment or care for a mental disorder (in particular section 3, section 7 (Guardianship), section 17A (Supervised Community Treatment) and forensic sections under Part 3 of the Act). It means that if the use of longer-terms forms of compulsion are being considered solely on the basis that a person has a learning disability, that disability must also be associated with abnormally aggressive or seriously irresponsible conduct. This does not, of course, preclude the use of compulsion for people who have another form of mental disorder (such as a mental illness) in addition to their learning disability.
Key Change 2: The Appropriate Medical Treatment Test
The MHA introduces a new “appropriate medical treatment” test that will apply to longer-term powers of compulsion concerned with treatment(for example, section 3 and Supervised Community Treatment ). As a result, it will not be possible for patients to be compulsorily detained for treatment or compulsion continued unless medical treatment which is appropriate taking into account the nature and degree of the patient’s mental disorder and all other circumstances of the case is available to that patient.
“Medical treatment” includes psychological treatment, nursing, and specialist mental health habilitation, rehabilitation and care as well as medicine. It does not have to be the “perfect” treatment, but doctors will be expected to satisfy themselves that appropriate treatment, taking into account all the circumstances of the case, is available and state in their recommendations in which hospital(s) it will be available to the patient. When making recommendations for section 3, the recommending doctors will need to confirm that there will be appropriate medical treatment available, and state on the recommendation form the hospital, hospitals or part of a hospital that the treatment will be given in.
Example:
Ellie was seen by 2 doctors in A&E after having been found by the police naked and dancing in the street. She was sexually disinhibited, and aggressive both physically and verbally. Ellie was well- known to the AOT team and was assessed by her own consultant and GP. Because they both knew her well, and knew what treatment would be of benefit to her, they felt confident in signing the form saying that appropriate medical treatment would be available, However, given her current presentation, they did not feel it would be safe to admit her to an open, mixed sex ward. Instead, her consultant rang and negotiated with the bed manager that she be admitted to a women only PICU bed. The ward name and hospital name were inserted onto the section 3 recommendation form, and the AMHP was only able to admit Ellie to that ward.
Key Change 3: Admitting young people to suitable environments
The effect of this change is that hospital managers are placed under a duty to ensure patients under 18 who are admitted to hospital for assessment or for treatment under the legislation, or who are voluntary patients are in an environment that is suitable for their age (subject to their needs). There is flexibility in the amendment to allow for patients under 18 years to be placed on adult psychiatric wards where the patient’s needs are better met this way. This is expected to come into force in 2010, by which time it is hoped new services will be available.
Section 140 of the existing Mental Health Act has also been amended to put a duty on Primary Care Trusts to let Local Social Service Authorities know where services especially suitable for admitting young people are to be found. The amendment to section140 has already come into force.
It is also now no longer possible to admit a competent and objecting 16 or 17yr old to hospital for treatment for mental disorder on the say of their parents.
Key Change 4: The Broadening of access to professional roles
This change widens the group of practitioners able to train to fulfil functions previously undertaken by Approved Social Workers (ASWs) and Responsible Medical Officers (RMOs). It does this by introducing two new roles:
Approved Mental Health Professionals (AMHPs): AMHPs are mental health professionals with specialist training in mental health assessment and legislation. The training will be opened up to include mental health and learning disability nurses, clinical psychologists and occupational therapists as well as social workers. AMHPs will assess “on behalf of” Local Authorities, who will continue to be responsible for approving AMHPs and for ensuring a 24hr AMHP service is available.
The final part of this change concerns the Approved Clinician (AC), the professional status/qualification a practitioner must obtain before they can take on the responsibilities of a Responsible Clinician (RC) for a particular person.
The RC is the old Responsible Medical Officer role which has now been opened up to include social workers, mental health and learning disability nurses, clinical psychologists and occupational therapists. The RC has overall responsibility for a patient’s case. This change allows more flexibility – for example, making it possible to transfer responsibility to professionals from different groups of staff as the patient’s needs change.
Directions make it clear that all professionals who want to be a RC need to meet particular levels of competence, undertake a short course to demonstrate their state of readiness and be approved by Strategic Health Authority as an AC.
Transitional arrangements for doctors:
Doctors with Section 12 status: for doctors with section 12 status, that status will continue unaltered for as long as had been expected to. For example, a doctor whose section 12 approval was due to expire in April 10, this will still be the case.
Doctors with section 12 status who have acted as an RMO for a patient in the 12 months prior to 3rd Nov 2008: such doctors will automatically be given Approved Clinician Status so that they can be Responsible Clinicians for people detained under the Act. If their section 12 status was due to expire during the next 12 months, it will be extended until 2rd Nov 2009.
Doctors (such as community consultants) who are section 12 approved, have been responsible for a patient’s medical treatment but have not acted as an Responsible Medical Officer (RMO) in the 12 months prior to 3rd Nov 08: such doctors will need to complete a course for Approved Clinicians before 2nd Nov 09, and as long as they do so their approval will last for a full 3 year period until 2nd Nov 2011.
In the future, a doctor (but not a professional from one of the other 4 eligible groups) who completes an Approved Clinician Course will also automatically receive section12 approval.
Accessing Approved Clinician Training:
AC training will be the responsibility of Strategic Health Authorities. Generally, people wishing to be Approved Clinicians will need to be recommended by employers – and will need to be able to demonstrate how they meet the competences for the role prior to undertaking the AC training course. The course is likely to be a few days at most, so preparatory training will be important. Each area will need to develop its own systems and policies, these are likely to be influenced by the pilot studies that have been taking place around the country in preparation for this change in the law.
Responsibilities of the Responsible Clinician (RC):
Renew detention with the agreement of another professional from a different professional background
Discharge patients from detention
Grant leave for patients.
Discharge patient on to supervised community treatment, with the agreement of an amhp
Set conditions on the community treatment order for sct with the agreement of an amhp
Extend sct with the agreement of an amhp
Recall sct patients to hospital, if necessary
Re- detain an sct patient by revoking the community treatment order, with the agreement of an amhp
Discharge patients from sct
Key Change 5: Nearest Relative Changes
Changes give patients the right to make an application to court to displace their nearest relative and introduces a new ground for displacement: that the current Nearest Relative(NR) is “otherwise unsuitable for the role”. The provisions for determining who is the NR have also been amended to include civil partners on equal terms with a husband or wife.
Key Change 6: Independent Mental Health Act Advocate (IMHA) service
Gives the right for patients who are subject to compulsion to have access to advocacy services. (that is, those on section 2, 3, section 7 Guardianship, SCT and similar forensic sections. Those on short term sections such as section 136 will not be eligible) Advocates will have the right to meet with patients in private. They will also have access to patient records, where a patient with capacity gives consent, and to meet and discuss the patient with professionals, such as doctors, involved in their care. In the case of patients lacking capacity to make such decisions, access must not conflict with decisions made by a deputy, Lasting Power of Attorney (LPA) donee or Court of Protection, and the person holding the records must agree that such access is “appropriate”. The principles of the COP should be used to decide whether it is appropriate to disclose information in a particular case.
It is planned that the new “Independent Mental Health Advocacy” services will be available from April 2009.
Key change 7: ECT and Patient Rights
Except in emergencies, detained patients may in future only be given ECT if they have capacity and agree or, (as now) if they do not have capacity, the ECT is authorised by a Second Opinion Appointed Doctor (SOAD).
In other words, this means that a detained patient can refuse to have ECT, and, except in emergencies, this can be overturned only if a SOAD agrees that the patient does not have capacity to make the decision and that giving the ECT treatment would be appropriate. In this case, the SOAD also needs to be sure that there is no valid advance decision refusing the use of ECT. If such an advance decision has been made, then ECT cannot be given, except in an emergency.
In the case of young people (aged under 18), even if the patient agrees, unless it is an emergency, they may only be given ECT with the additional agreement of a SOAD. These rules apply to young people whether or not they are detained.
In all these cases, it is only an emergency if the ECT is immediately necessary to save the patient’s life or prevent serious deterioration in their condition.
Key Change 8: Supervised Community Treatment
Introduces Supervised Community Treatment (SCT) for patients following a period of detention in hospital for treatment (mainly those on section 3 or unrestricted forensic sections such as section 37). It will allow a small number of patients who currently disengage from support once discharged from hospital to be cared for in the community, subject to the possibility of being recalled to hospital if necessary. This is to ensure they continue to get the treatment they need. There is no lower age limit.
As a statutory framework, SCT is intended to support such vulnerable patients (including some who may pose a risk to others) to:
live in the community;
help improve engagement with the care team by shifting the balance of power more in the patient’s favour;
act upon any clinical signs of relapse at an early stage;
be a mechanism to manage actual or potential relapse; and
ensure that services are aware of and responsive to any changes of circumstances which arise for the patient or their carers.
The criteria for consideration of the use of SCT include:
the person is suffering from a mental disorder and detained under s3 (or similar forensic section) for treatment;
the need for medical treatment;
the existence of a risk to the patient’s health or safety or that of others;
that appropriate treatment is available; and that the patient does not need to be in hospital to receive it but does need to be liable to recall to hospital to ensure that the risk can be managed; and
that it is necessary for the patient’s health or safety or the protection of others that the patient remains liable to recall.
Key Change 9: Changes to the Tribunal system
Changes to the MHA have introduced earlier referrals by Hospital Managers of detained patients who have not used their rights of appeal to the Tribunal.
To protect patients, everyone who is compulsorily admitted to hospital must have their detention review by the Tribunal service within the 1st 6 months, and every 3 years after that. If the patient themselves doesn’t request a tribunal referral within the above timescales, the hospital managers must do so.
For under 18s, they must be seen annually by the tribunal.
The Secretary of State has the power to reduce further these periods for referral by Hospital Managers in the future.
The MHA 2007 has also introduced the immediate referral of patients who have had their SCT revoked.
The structure of the Tribunal Service has been changed to provide a ‘1st’ and ‘2nd’ tier. The purpose of the 2nd tier is to allow appeals and clarification of legal points, without having to use the judicial review process.
In addition, new rules have been issued as guidance for professionals about what is expected to be covered in reports to tribunals.
Key Change 10: the role of the 2nd Professional in the renewal of section 3
In addition to the Responsible Clinician, the written agreement of a second professional is needed to renew section 3.
The second professional must
come from a different professional discipline compared with the RC
be involved with the care of the patient
be able to reach independent decisions
have sufficient expertise and experience to make a judgement about whether the criteria for detention continue to be met.
Even if the RC disagrees with the view of the 2nd Professional, they should not normally seek to find another professional. In such circumstances the section 3 would not be renewed. In very exceptional circumstances where a different opinion is sort, this must be brought to the attention of the hospital managers who are also required to consider and approve the renewal of the section.
ACKNOWLEDGEMENTS
This article is based on work undertaken as part of the NIMHE Implementation team for the Mental Health Act, and forms part of the National Training Materials available to all staff working within Mental Health
COMPETING INTERESTS
None Declared
AUTHOR DETAILS
CLAIRE BARCHAM, National Coordinator, AMHP leads Network/ NIMHE National Implementation team member,United Kingdom.
CORRESPONDENCE: CLAIRE BARCHAM, ASW Training and Development Coordinator, C&I MHSCT Training and Development Team, Room 110, West Wing, St Pancras Hospital, 4 St Pancras Way, London NW1 0PE
Email: claire.barcham@candi.nhs.uk
For more information, please visit https//:mhact.csip.org.uk
Obesity has become a ticking time bomb. The population of obese people and so also obese pregnant patients is increasing worldwide and it won’t be long before when anaesthetists will be more commonly faced with managing obese parturients with a large spectrum of comorbidities. The last confidential enquiry into maternal and child health (CEMACH) 2002 - 2005 report stressed obesity as a major risk factor associated with maternal mortality and following suit of its recommendation, we write this review article on management of obese parturients, highlighting the problems in obese parturients and recommending guidelines for management of such patients. As the use of regional anaesthesia in obstetrics anaesthesia has increased, the trainee anaesthetists are relatively less skilled to provide general anaesthesia. General anaesthesia with all the airway management problems has been the major reason of maternal mortality in the previous CEMACH reports. An epidural block though technically difficult, provides optimal analgesia and can be extended for caesarean section if required. Hence obese parturient should be assessed and consulted by a senior anaesthetist as early as 28 weeks of gestation in the pregnancy for formulating a plan for labour analgesia and anaesthesia for caesarean section if required. Epidural analgesia should be provided in early labour prophylactically to avoid general anaesthesia. Early anaesthetic assessment, prophylactic epidural block, ensuring its effectiveness, alternative plan for failed regional block along with preparation for general anaesthetic and difficult intubation, involving senior help in the management and multidisciplinary approach are advocated to mitigate potential anaesthetic risks.
Abbreviations: BMI - Body mass index
Obesity has become a major health problem of modern society and increasing globally at nearly epidemic proportions especially in western and European countries 1,2,3,4.
DEFINING OBESITY
Obesity can be simply defined as a condition in which body fat is in excess beyond a point incompatible with physical and mental health and normal life expectancy 5 or as a metabolic disorder that is primarily induced and sustained by an over consumption or underutilization of caloric substrate. There are 2 types of obesity; Android obesity which is truncal distribution of fat associated with high incidence of cardiovascular disorders and Gynecoid obesity where fat is distributed to thighs and buttocks associated with pregnancy and not tightly linked to cardiovascular problems 6, 7.
Indices used to for obesity are Ideal body weight in kilograms (Broca’s Index), and more commonly the BMI or body mass index
( also called Quetelet’s index ).
1) Ideal body weight = height in centimeters - 100 for men ( 105 for women ). Overweightness is 20% more than ideal body weight and morbid obesity is twice the Ideal Body weight.
2) BMI = weight in kgs/ square of height in meters
Prevalence
In the US more than 60 million adults can be classified as either overweight or obese with morbid obesity affecting more than 9 million adults. Approximately 30 – 40 % of females are obese and it is estimated that 50 per cent of women will be obese by 2050. A study looking at trends in pre-partum obesity in nine states of the United States found an increase in pre-partum obesity from 13% in 1993–1994 to 22% in 2002–2003 8.
WHO classification of Obesity 9
Classification
Body mass index (kg/m2)
Associated health risks
Underweight
<18.5
Low
Normal range
18.5–24.9
Average
Overweight
>25.0
Preobese
25.0–29.9
Increased
Obese class I
30.0–34.9
Moderately increased
Obese class II
35.0–39.9
Severely increased
Obese class III
>40
Very severely increased
In the UK, 56% of all women are over the recommended BMI, with 33% of them classified as overweight (BMI > 25) and 23% obese (BMI > 30). The Health Survey of England published in 2002 gives data about the prevalence of obesity in England. Females in the reproductive age group (16 – 44 years) have shown a dramatic increase in BMI. The percentage of women with BMI above 30 increased from 12% in 1993 to 18.3% in 2002. Also alarming is that the percentage of morbidly obese women has doubled in the last decade 10. The dramatically increasing rate of obesity in the general population also extends to women of reproductive age.
Pathophysiological changes in obese pregnant patient
Obesity compounds most of the physiological changes in pregnancy
Airway - Obesity and pregnancy each increase the chance of difficult airway. Limited mouth opening and limited neck movements are common in obesity. There is narrowing of the pharyngeal opening due to excess adipose tissue and on airway examination, the airway will have more commonly high of mallampati grades. In pregnancy, particularly in pregnancy induced hypertension, the mucous membranes in the airway are oedematous and hence more prone to bleeding. Breast enlargement in pregnancy also predisposes to difficult airway.
Respiratory system – There are significant changes in an obese parturient and most of them are additive. In early pregnancy , in a non obese parturient, even before the uterus is large enough to affect respiratory function, women begin to have a sensation of dyspnea. This sensation likely occurs from the increased alveolar ventilation, probably secondary to progesterone effects on the respiratory center in the brainstem 11,12. By the fifth month of pregnancy, the growing uterus begin to cause a progressive decrease in expiratory reserve volume (ERV), residual volume (RV) and functional residual capacity (FRC), which at term are about 15–20% less than those of the non-pregnant state13. Obesity in non-pregnant subjects is associated with a decrease in expiratory reserve volume (ERV), residual volume (RV) and functional residual capacity (FRC), most likely caused by the added weight of excess fat on the chest and abdomen and decreased chest compliance13 – 16. Eng et al.13 showed, however, that obese parturients did not have a significant additional reduction in functional residual capacity (FRC) compared to normal-weight parturients, which might be partially explained by the fact that the study was performed with the parturients in the sitting position. Another possible explanation is progesterone which has a relaxing effect on smooth muscle and decreases airway resistance, thus reducing some of the negative effects of obesity on the respiratory system 12, 17.
Dempsey et al.18 have showed that excess body weight in obesity increases oxygen consumption and CO2 production in a linear fashion. The work of breathing is increased in obese parturients due to chest wall weight and they typically show a rapid and shallow breathing pattern 18. This leads in turn to a higher ventilatory requirements and work of breathing 12,19. The supine, lithotomy, induction of general anaesthesia and especially the Trendelenburg position worsen lung volumes significantly. The funtional residual capacity (FRC) is further reduced and the closing capacity (CC) encroaches on the funtional residual capacity (FRC) resulting in small airway collapse, ventilation perfusion mismatch, shunting and hypoxemia20. These physiologic changes make the obese parturient particularly prone to rapid desaturation, stressing the importance of adequate denitrogenation ('pre-oxygenation') before induction of general anaesthesia.
In non-obese parturients, physiologic changes during pregnancy are thought to protect from obstructive sleep apnea, due to high circulating levels of progesterone, which is a ventilatory stimulant 18. However, obesity increases the risk for obstructive sleep apnea significantly and this syndrome is not uncommon in the obese parturients. Obesity hypoventilation syndrome (OHS , Pickwickian syndrome ) is seen in 8% of population of obese parturients characterized by morbid obesity, alveolar hypoventilation and daytime somnolence. In response to chronic hypoventilation and hypoxemia, they develop polycythaemia, increased cardiac output, cardiomegaly, pulmonary hypertension and eventually right heart failure. There is a significant increase in morbidity and mortality. They are more to obstructive sleep apnoea. Pulmonary embolism and pneumonia are also common in these patients 25.
Cardiovascular system – Cardiac output increases in pregnancy, with a significant increase in cardiac output, becoming detectable by the third week of pregnancy and a 35 - 40% increase by the end of the first trimester. Cardiac output continues to rise throughout the second trimester until it reaches a level that is approximately 50% more than that in the non-pregnant state. For the remaining pregnancy, cardiac output remains relatively stable around that level. During labour, cardiac output increases further by approximately 10% in the early first stage, 25% in the late first stage and 40% in the second stage. Uterine contractions increase cardiac output by further 10–15% and in the immediate post-partum period the cardiac output peaks at as much as 75% above prepartum values 19. Obesity increases cardiac output even further because of extra amount of fat. Every 100 g of fat increases the cardiac output by 30–50 ml/min22. Blood volume is increased in pregnancy and even more when pregnancy is complicated by obesity. In non-obese parturients, there is a significant reduction in afterload 22. In obese pregnant parturients, however, afterload reduction may be impaired due to increased peripheral resistance and greater conduit artery stiffness 23. Additionally, obesity is associated with a higher prevalence of hypertension, diabetes mellitus, hyperlipidemia and poor cardiac function and it is one of the leading risk factors for coronary artery disease and cerebrovascular accidents 24. Due to hyperdynamic circulation, there ensues left ventricular hypertrophy and diastolic dysfunction. Systolic function might remain normal but progressively systolic dysfunction may ensue. Pulmonary blood volume increase due to increased cardiac output. Pulmonary hypertension can develop and is exacerbated by supine position, airway obstruction and hypoxemia can develop. In obesity hypoventilation syndrome, right ventricular failure can develop. Increased number of peripartum cardiomyopathy cases are seen in obese pregnant parturients but it is unclear if obesity is a risk factor 25.
The obese pregnant parturients are at an increased risk of supine hypotension syndrome (SHS) due to compression of major abdominal vessels. This is exacerbated by large panniculus which adds to the uterine compression. Tseuda et al. have reported two cases of sudden death on assuming the supine position in morbidly obese patients 26.
Gastrointestinal system – Obesity further decreases lower oesophageal tone which is already decreased in pregnancy and increase the risk of aspiration of gastric contents and Mendelson’s syndrome 27, 28. Hiatus hernia is increased in obese patients. Roberts and Shirley studied obese and non obese pregnant parturients in labour; the gastric volume in obese parturients is five times greater than in the controls 29,30,31. Obese population have a higher incidence of diabetes, which can cause delayed gastric emptying, increasing the risk for aspiration. Also, it is well known that obesity predisposes to difficult or failed intubation, both of which are associated with a higher incidence of aspiration.
Others - Gestational diabetes is common. Obesity metabolic syndrome includes dyslipidemia, impaired endothelial function, high blood pressure, increased inflammatory mediators, insulin resistance and hyperinsulinemia even in absence of diabetes 25.
Pharmacokinetics and pharmacodynamics changes
Obesity increases both fat and lean masses; however, the percentage of fat tissue increases more than does the lean mass, affecting the apparent volume of distribution of anaesthetic drugs according to their lipid solubility. Thiopental sodium and propofol dosages are calculated on total body weight (TBW). Benzodiazepine loading doses should be adjusted on actual weight, and maintenance doses should be adjusted on ideal body weight. The loading dose of lipophilic opioids is based on total body weight (TBW), whereas maintenance dosages should be cautiously reduced because of the higher sensitivity of the obese patient to their depressant effects. Pharmacokinetic parameters of muscle relaxants are minimally affected by obesity, and their dosage is based on ideal rather than total body weight (TBW). Minimum alveolar concentration is decreased. Inhalation anaesthetics with very low lipid solubility, such as sevoflurane and desflurane, allow for quick modification of the anaesthetic plan during surgery and rapid emergence at the end of surgery, hence representing very flexible anaesthetic drugs for use in this patient population. Drug dosing is generally based on the volume of distribution for the loading dose and on the clearance for maintenance. In the obese patient, the volume of distribution is increased if the drug is distributed both in lean and fat tissues whereas the anaesthetic drug clearance is usually normal or increased 32. Albumin binding of drugs is unchanged in the obese, but levels of fatty acids, triglycerides, and a1-acid glycoprotein are increased and may influence plasma protein binding. In pregnancy, the volume of distribution is increased, albumin concentration decreased and the renal clearance is increased. Net effect is unpredictable. Pseudocholinesterase levels are decreased in pregnancy.
Local anaesthetic requirements
Lower dose of local anaesthetic is required (less by 25%) when injected neuraxially. Proposed mechanisms are pregnancy induced hormone related changes in the action of spinal cord neurotransmitters, potentiation of the analgesic effect of the endogeneous analgesic systems, increased permeability of the neural sheath 25 and decreased dilution by decreased volume of cerebrospinal fluid (CSF) Hodgkinson et al 33 have shown an increased cephalad spread of local anesthetics in obese patients. Hogan et al.34 found a lower average cerebrospinal fluid (CSF) volume in obese subjectsI, which could explain the decreased local anesthetic dose requirements due to decreased anaesthetic dilution. Since similar changes were noticed with external abdominal compression and abdominal pressure increases linearly with increased body weight 35, increased abdominal pressure is probably the cause. Some 36 have also attributed the decrease in cerebrospinal fluid (CSF) volume to compression of the dural sac due to engorgement of the epidural venous plexus and increased epidural space pressure, resulting from compression of the inferior vena cava by gravid uterus with redistribution of the venous return from the lower limbs and pelvis. Hogan et al.34, however, suggested that the mechanism by which increased abdominal pressure decreases the CSF volume is probably inward movement of soft tissue (mostly fat) in the intervertebral foramen, which displaces CSF. This hypothesis is based on their findings that the greatest change in CSF volume during abdominal compression was found at sites in which intervertebral foramina were present. Greene suggests that larger buttocks of obese patients place the vertebral coloumn in the Trendelenburg position, exaggerating the cephalad spread of the local anaesthetic 25, 37
Consequences of obesity in pregnancy
Obesity severely complicates pregnancy. It affects both the mother and foetus
Maternal consequences
There is increased risk of gestational diabetes and type 2 Diabetes. There is 3 - 10 fold higher risk of gestational diabetes (type 1 insulin dependent diabetes mellitus)38. Studies have shown when pregnancy is complicated by gestational diabetes, there is a higher risk of developing type 2 diabetes mellitus in later life 39. There is a 2 – 4 fold increased risk of preeclampsia. The risk is almost 5 times greater in the morbidly obese group; typically a BMI > 35 38, 40. But there is no increased risk of HELLP (haemolysis, elevated liver enzymes, low platelets) syndrome 31. Obesity is an independent risk factor for hypertension 41.
It is reported 42 that there is a higher chance of failure to progress, prolonged second stage of labour and a failed induction of labour in obese compared to non obese parturients and this is secondary to soft tissue dystocia. There is a higher risk of instrumental delivery of up to 18% in women with a BMI between 35 and 40 and up to 34% in patients with BMI greater than 40. Also there is an increased risk of failed instrumental delivery leading to caesarean section. There is 3 times higher risk of caesarean section in a obese parturient. This is due to fetal macrosomia, higher risk of shoulder dystocia and/or failed cervical dilatation 38, 43, 44. About two thirds present as emergency caesarean section 45. The obese parturient is at a higher risk of having a prolonged incision to delivery time, blood loss greater than 1000 ml and prolonged operative times. There is an increased risk of wound infections and endometritis and dehiscence 46, 47. There is an increased risk of major postpartum haemorrhage. The risk of postpartum haemorrhage rises with increasing BMI and is about 30% more frequent for a moderately raised BMI and about 70% more frequent for a highly raised BMI compared with the normal BMI group 45,48. There is an increased risk of thromboembolism – obesity and pregnancy are each independent risk factors for deep vein thrombosis. Both pharmacological and mechanical methods should be used for thromboprophylaxis.
Obese women spend an average of 5 more days in hospital resulting in 5 fold increase in cost of care due to potential complications such as wound infections and postpartum haemorrhage 49.
Fetal consequences
Maternal obesity is associated with large for gestational age infants. There is increased risk of a macrosomic foetus, independent of maternal diabetes 43, 50. There is an increased risk of shoulder dystocia upto three times more common in the morbidly obese parturients. The risk of foetal macrosomia and shoulder dystocia increases with increase in BMI 44.
There is an increased risk of Infant birth defects. Since 1994 a number of studies have shown an association between maternal obesity and infant birth defects. Anomalies include neural tube defects such as anencephaly, anomalies of the heart and intestinal tract, omphaloceles, orofacial clefts, and multiple congential anomalies of the central nervous system ( 43, 51, 52 ).
There is an increased risk of stillbirth, a three times increase in antepartum stillbirth was found in morbidly obese parturients compared with women of normal BMI 52, 53.
Due to the depth of maternal adipose, foetal monitoring by intermittent or continuous Electronic Foetal Monitoring using external transducers may be technically difficult. The use of fetal scalp electrodes and intrauterine pressure catheters to ensure an acceptable standard of fetal monitoring may be needed.
In a study ‘Maternal obesity and pregnancy outcome: a study of 287213 pregnancies in London’ (N J Sebire, et all, International journal of obesity (2001) 25, 1175 - 82 ) complications such as gestational diabetes mellitus proteinuric pre-eclampsia; induction of labour; delivery by emergency caesarean section; postpartum haemorrhage; genital tract infection; urinary tract infection, wound infection; birth weight above the 90th centile, and intrauterine death were significantly higher in obese pregnant parturients than non obese pregnant parturients. However, delivery before 32 weeks gestation and breastfeeding at discharge were significantly less likely in the overweight groups. In all cases, increasing maternal BMI was associated with increased magnitude of risk 38. Weiss et al.54 found for nulliparous patients a caesarean delivery rate of 20.7% in the control group compared with 33.8% in the obese and 47.4% in the morbidly obese group.
The Confidential Enquiry into Maternal and Child Health 2004 reported that 35% of all maternal deaths occurring in the triennium 2000–2002 were in obese women with BMI > 30. The most recent CEMACH reports in the United Kingdom reported that obesity was a cofactor in a significant number of the maternal deaths between 2003 and 2005. Twenty seven percent of the women who died had BMI > 30. Of these women, 12% had a BMI between 30 and 34, 7% had values between 35 and 39 and 8% had a BMI of 40 or more. 295 women who died 119 were overweight and 64 of those were morbidly or super-morbidly obese. In 30 per cent of women who experienced a stillbirth or perinatal death, the maternal BMI was recorded at more than 30.
Furthermore, Cedergren et al found a 3 fold increased rate of stillbirths, 5 fold increased risk of preeclampsia and a 3 fold increased risk of caesarean section. The success rate for a vaginal delivery in obese parturient with a previous caesarean section is less than 15% 38.
Anaesthetic management
Obese parturients have severely limited physiological reserve and a higher risk of emergency surgical intervention. Hence the anaesthetic risks increase greatly. Obesity and pregnancy each has multisystem effects, many of which are additive. A thorough understanding of the physiology, associated conditions and morbidity, available options for anaesthesia and possible complications is important.
Senior anaesthetist must be involved early in multidisciplinary approach for patient care as early as 28 weeks of gestation. The preoperative assessment include evaluation of airway, respiratory and cardiovascular system and pregnancy associated problems such as pregnancy induced hypertension, gestational diabetes etc and also should include patient education. An examination of the back should be done.
Airway
The obese parturients need thorough pre-operative assessment for difficult airway as incidence of failed intubation is 8 times higher than non obese patients. In the obstetric population, between one in 280 and one in 750 attempted tracheal intubations fail 45, compared to one in 2230 in the general population 17, 55,56. In contrast, the incidence of difficult intubation in obese population, is as high as 15.5% 57. Dewan 58 found the incidence as high as 33% in morbidly obese parturients. A 6-year review of failed intubations in parturients in a United Kiingdom region reported 36 cases of failed intubation and it was found that the average BMI of these women was 33 57. So it is evident that incidence of difficult or failed tracheal intubation in obese parturients is very high and emphasizes optimal assessment and management of the airway. An airway assessment should include mallampati classification, thyromental distance, neck extension (atlanto-occipital joint extension), mouth opening (vertical dimension). The combination of two tests (mallampatti and thyromental distance), though in a small study of 80 parturients receiving general anaesthesia, has been shown to be 100% sensitive with 70% positive predictor value 59. These tests can be done in less than 1 minute; hence they are also useful in an emergency scenario. Other features shown to be of significance are short neck, receding mandible and protruding incisors 60. It is of interest to note that neck circumference, not BMI, is more predictive of a difficult intubation in morbidly obese patients 61. A study has shown a gestational weight gain of more than 15 kgs is associated with three times increase in suboptimal layngoscopic view as compared to that in non obese parturients of the same age 57, 62. This means weight gain in pregnancy should be limited in obese parturients and if an obese parturient presents who has gained more than 15 kgs of weight in pregnancy, she will be more likely to have a difficult airway 62. A plan of airway management should be formulated in case of an emergency for all women regardless of the primary obstetric and anaesthetic plan. Although rapid sequence intubation with proper positioning and back up equipment may be adequate for most women, an alternative airway plan should be considered. A history of snoring, diagnosis of sleep apnoea, lack of teeth, and large breasts all increase risk of difficult intubation and awake fibreoptic intubation should be considered in all patients with limited range of neck, head or jaw movements, short neck, neck circumference of 15 inches and above, and mallampati score of 3 and above 25
Respiratory System
Usually a complete history and chest examination and routine investigations including an ECG is adequate for a preoperative anaesthetic fitness. However chest X ray, arterial blood gas, pulmonary function tests can be done to aid further evaluation of respiratory reserve. Measurement of oxygen saturation by pulse oximetry in sitting and then supine can provide evidence of airway closure during normal tidal volume ventilation, thereby identifying candidates for post operative oxygen administration 17.
Women with obesity are more likely to have obstructive sleep apnoea but the prevalence is unknown in pregnancy. Sleep disturbances and day time fatigue are normal at the end of pregnancy and so obstructive sleep apnoea may go undiagnosed. J Mhyre 25 has suggested women with a BMI > 35, neck circumference of greater than 16 inches, symptoms of suspected airway obstruction during sleep ( include frequent or loud snoring, observed pauses in breathing during sleep, frequent arousals from sleep or arousal with a choking sensation ) should be screened by polysomnography for obstructive sleep apnoea and advised continuous positive airway pressure (CPAP) if required.
If obesity hypoventilation syndrome is suspected arterial blood gas is useful to screen hypoxia, hypercarbia and acidosis and echocardiogram should be done to evaluate cardiac function and patient should be referred to cardiologist 25,63
Cardiovascular system
Cardiovascular co-morbidities such as hypertension, ischaemic heart disease and heart failure can co-exist in obese parturients. Nearly 40% of the obese population experience angina without demonstrable coronary artery disease 64. Pulmonary hypertension can be present. Hence cardiologists should be involved early in the care of symptomatic morbidly obese parturients to investigate and optimise the disease status wherever appropriate 17. Echocardiogram may be useful.
The obese parturients cannot be accurately stratified for perioperative risk using the usual screening indices such as Goldman’s index etc as obesity and pregnancy are not included as risk factors in these indices and they might be classed in the lower risk group despite having significantly increased risk.
Others
Patients should be assessed for pregnancy associated problems such as pregnancy induced hypertension and gestational diabetes mellitus etc.
Peri-operative issues such as transfers, beds, intravenous access, central venous access, difficulty in measuring non invasive blood pressure, arterial cannulation, different size regional anaesthesia kit should be anticipated, discussed and planned for.
Post operative intensive care management /high dependency care should be sought for. Deep vein thrombosis prophylaxis must be put in place. The management plan should be liaised with whole team including consultant anaesthetists, consultant obstetricians, consultant intensivists, midwives, operating department practioners (ODP’s) and physiotherapists
Analgesia for labour
Each of the risk factors of fetal macrosomia and shoulder dystocia which are increased in obese parturient result in more painful contractions and complicated labour 65. Although there are various modalities of pain relief, analgesia using neuroaxial blockade has been shown to be the most effective 66. The anticipated technical difficulties should not preclude the use of epidural analgesia in obese parturients. It is been shown effective pain relief during labour can improve maternal respiratory function and attenuate sympathetically mediated cardiovascular responses 67, 68. Available evidence shows that the rate of caesarean delivery does not increase with epidural analgesia during labour 66, though obesity increases the need for caesarean section. Hence, placing a functional epidural catheter is advantageous should any operative intervention be required. In addition, epidural analgesia can be extended into the postoperative period where adequate pain relief can optimise care.
The challenges for the anaesthetist should not be underestimated. Technical problems include appropriate positioning of the patient, identification of the midline and epidural space, and dislodgement of catheters 45, 69, 70. The initial failure rate for epidural catheter placement can be very high (42%) 45 and multiple attempts of catheter placement are common. Jordan et al. noted 74.4% of massively obese parturients needed more than a single attempt and 14% needed more than three attempts for successful epidural placement 71. The knee–chest position required for doing epidural in the lateral position is difficult to obtain in the obese. One study has shown that cardiac output decreased more in the lateral decubitus position with maximal lumbar flexion compared with the sitting position 72. Moreover, in the lateral position, gravity can drag down the pad of fat obscuring the midline. Another study found the depth of the epidural space from skin to be greater in patients where the epidural was inserted in the lateral decubitus position 73. Overall, the sitting position is preferable and should be used.
Steps and caveats
Early placement and confirmation of optimal epidural analgesia even before onset of labour ( when a term patient presents before labour ) is prudent. This allows sufficient time to manage a failed epidural block ( because not only the incidence of failed initial epidural catheter placement is high in obese parturients, but the incidence of failed epidural during labour due to migration of epidural catheter in the fatty subcutaneous tissues is also high )17, 74, 75. Re-evaluate the airway and cardiorespiratory status. Senior anaesthetist preferably a consultant anaesthetist should be involved. Ensure wide intravenous cannula (preferably 14 or 16 gauge) in place. In case of problems with blood pressure cuff not measuring, cuff can be placed on calf/forearm; will help in getting the trends if no accurate reading. Invasive blood pressure monitoring might be needed. Ensure pulse oximetry monitoring and supplement oxygen by mask if required.
Perform in sitting position. Ensure midline position as even if slight deviation of the midline will lead to exaggerated directional errors due to increased length of epidural space from the skin and hence failure of epidural. Midline might be not possible to palpate, in this case drop a line from C1 spinous process to lower skin crease and this may be guide as a midline. Strapping excess fat away from the midline might be necessary.
If highest points of iliac crests are palpated for the Tuffier’s line then because of fat pads on the sides, higher spaces might be inadvertently selected and increased chance of spinal cord damage. In case of difficulty, lower thoracic space may be selected.
A recent study in pregnant patients has shown a positive correlation between BMI and the distance to skin to the lumbar puncture 76. Although the epidural space may be deeper in overweight people, the majority of studies report that only a few have an epidural space deeper than 8 cms 73, 77. Hence it seems appropriate to use a standard needle to identify the epidural space on the first attempt. In morbidly obese patient ultrasound technique has been found valuable in establishing epidural 78, 79
In case of difficulty in insertion, a deliberate spinal with 25 guage needle might be performed ( no injection of drugs ) to assess the midline and depth of epidural space. There is an increased risk of dural tap 25, but decreased risk of postdural puncture headache 75. In case of dural tap, epidural can be converted to continuous spinal catheter analgesia with extreme caution. Also there is an increased risk of Intravenous placement of epidural catheter due to engorged epidural veins and decrease in epidural space. The meniscus drop ( negative pressure ) test is not reliable as epidural pressure may be high 25. Minimum 5 cms of catheter in space should be left. To minimize catheter displacement, it should be secured on assumption of upright or preferably lateral position from the initial flexed position. The epidural should be checked with a test dose and a functioning epidural should be ensured. Sometimes a longer epidural needle might be required. There is an advantage to titrate block height. Minimum local anaesthetic concentration ( MLAC ) is lower in obese pregnant patients compared to non pregnant patients 80
If epidural is contraindicated or impossible to site then entonox is an useful adjunct. Intramuscular opioids are not reliable. Patient controlled analgesia can be used but cautiously as increased chance of sedation and respiratory depression. Remifentanyl, an ultra short acting opioid, has favourable pharmacokinetics to be used as an opioid for patient controlled analgesia but not enough data is available for its use in obese parturients. It is metabolized by red blood cells and tissue esterases both in mother and foetus and hence does not accumulate and is easily antagonized if required. However it is a potent respiratory depressant and hence should be used very cautiously in obese parturients who would be susceptible to its sedative side effects and hence they should be managed in high dependency unit with appropriate monitoring and one to one nursing by skilled midwife and under observation of a highly skilled anaesthetist. The dosage should be carefully titrated individually and naloxone and difficult airway trolley ready. Patients with obstructive sleep apnoea would be very susceptible to its sedative side effects and hence should be avoided. Proper training of patients is required as its peak effect is 2 -3 minutes and if the button of patient controlled analgesia is pressed at the onset of contraction it would be less effective. The duration of its use should be minimized as much as possible.
Analgesia/anaesthesia for Caesarean section
Obesity and Caesarean section have been identified as independent risk factors for maternal morbidity and mortality 44. Analysis of direct maternal deaths due to anaesthesia, in the confidential enquiries report on maternal mortality in the United Kingdom from 1979 to 2005, reveals that the majority of deaths occurred under general anaesthesia, compared with regional anaesthesia 17. Most parturients who die of complications of general anaesthesia die of airway management problems, including aspiration, failed intubation, inadequate ventilation, and respiratory failure.54, 55
Factors that play a role in general anaesthesia being more likely to be associated with maternal mortality than regional anaesthesia are unexpected airway difficulties, pulmonary aspiration of gastric contents, emergency general anaesthesia (including conversion of a failed regional), peripartum haemorrhage, and embolism necessitating general anaesthesia, and resident lack of experience in general anaesthesia for caesarean section 28 , 81.
In Why Mothers Die 2000–02, 35% of all the women who died were obese, 50% more than in the general population 82.
Direct maternal deaths due to anaesthesia by types of anaesthesia in United Kingdom 1979–2005. Derived from CEMD reports. Since 1979, maternal deaths are reported as direct and indirect.
Year
Total(n)
GA(n)
RA(n)
Other (n)
2002-05
6
4
1
1
2000-02
6
6
0
0
1997-99
3
2
1
0
1994-96
1
0
1
0
1991-93
8
7
1
0
1988-90
4
3
1
0
1985-87
8
7
1
0
1982-84
18
17
1
0
1979-81
22
22
0
0
GA, general anaesthesia; RA, regional anaesthesia; Other – eg Central VP line insertion
Hence regional anaesthesia preferably epidural should be opted unless contraindicated or difficult.
Regional anaesthesia for Caesarean section
Different techniques can be used. Epidurals are reliable but have high failure rate, spinal is a familiar technique while combined spinal epidural has minimal side effects such as headache, high block , hypotension and can be used post operatively as well as for redo surgery.
Use 25% less local anaesthetic dose compared to non obese patient due to altered neuro-axial physiology and anatomy.
Epidural
A working epidural as above can be continued for caesarean section and it also provides post operative pain relief. However it may be inadequate in more than 25% of these patients, mainly because of difficulty in blocking the sacral roots, resulting in visceral pain upon stimulation of the bladder 83.
Spinal
An obese woman is a candidate for spinal anaesthesia if the airway examination is normal, cardiopulmonary derangements are minimal and the obstetricians aim to complete the surgery within 90 minutes 25. In obese parturients spinals can be technically difficult requiring varied needle lengths and being unable to titrate to block for surgery and surgical duration. If the spinal wears off, general anaesthesia with all its inherent risks, will be required. Tuohy needle can be used as an introducer for the spinal needle 25. Spinal opioids can provide post operative analgesia but respiratory monitoring becomes essential.
Combined Spinal Epidural
Success of combined spinal epidural will depend on familiarity with technique. It is more versatile to titrate the block and dose and also a faster onset compared to epidural alone. This technique can be useful for post operative analgesia and re-operative anaesthesia 84. There is higher rate of success for surgical anaesthesia compared to spinal or epidural alone. Several studies have shown that catheters inserted as a part of combined spinal epidural technique produce anaesthesia /analgesia more reliably than those inserted via a standard epidural technique 85 – 88. The appearance of cerebrospinal fluid indirectly confirms correct epidural needle placement and increase the chance of functional epidural catheter. There is a possible flaw when spinal injection alone produces the desired block and epidural remains untested; when epidural is required and fails, general anaesthetic might be needed 25 and hence a small dose intrathecally might be used to establish the analgesia to make mother pain free (which therefore also decreases the risk of hypotension) and then epidural should be used to make sure it is working for the complete surgical anaesthesia.
Continious Spinal anaesthesia
Operators need to be familiar with technique. Continuous spinal anaesthesia must be done always with consultant anaesthetist. It is occasionally used in patients who have accidental dural puncture. It may be used when epidural is indicated and difficult to site. It provides reliable and predictable block and allows to titrate the block to desired level and duration. It provides surgical anaesthetic level within minutes in emergency situations with incremental doses. It is important to flush the catheter before placement to avoid introducing air into the spinal space which could cause pneumoencephalus headache 25. It is also very important to mark it as an intrathecal catheter and to be used by anaesthetist only. This can be used for analgesia as well as anaesthesia.
Incidence of headache and infection is higher with this technique compared to other regional techniques but overall incidence of post dural puncture headache in obese parturients is lower 89, 90. Final density and level are proportional to the dose in mgs, not the volume delivered
General anaesthesia for Caesarean
Consultant anaesthetist should be involved as early as possible. Strategy should be to avoid need for emergency general anaesthesia by being proactive and establishing effective regional analgesia and anaesthesia as early as possible. Airway assessment regarding difficult airway must be done. Preparation for general anaesthesia and difficult intubation (ensure lower sized endotracheal tube and a laryngeal mass airway) must be in place including awake fibre optic laryngoscope. Anti-aspiration prophylaxis must be given before conduct of anaesthesia
Collins et al. 91 investigated the effect of the position of the patient on the view obtained during laryngoscopy in 60 morbidly obese patients. They found that the 'ramped' position, accomplished by arranging blankets underneath the patient's upper body and head until horizontal alignment is achieved between the external auditory meatus and the sternal notch, clearly improves the laryngeal view when compared with the standard 'sniff' position. HELP (Head elevated laryngoscopy position) should be given to make sure that airway is in alignment.
After all monitoring including foetal monitoring is in place, patient must be prepared awake and draped. Adequate preoxygenation { 8 vital capacity breaths of 100% oxygen 92} is ensured as otherwise they rapidly desaturate. Baraka et al. 92 showed that pre-oxygenation achieved by eight vital capacity breaths within 60 s at an oxygen flow of 10 liters/min not only results in a higher partial pressure of arterial oxygen (PaO2), but also in a slower hemoglobin desaturation when compared with the four deep breaths technique. Use standard rapid sequence induction with cricoid pressure and left lateral tilt in patients with no anticipated difficult airway. For general anaesthesia make sure drug doses for (thiopentone, suxamethonium, atracuruim) are calculated before hand keeping in view altered distribution and elimination in obese patients. Dewan suggests that at least 4 mgs/kg of thiopentone (up to a maximum dose of 500 mgs) should be used if chosen, to avoid the risk of maternal awareness, hypertension and decreased uterine blood flow during light anaesthesia 93. Administration of a larger dose may be associated with delayed arousal in the event of failed intubation. For suxamethonium, dose based on 1 - 2 mgs/kg of actual bodyweight up to maximum of 200 mgs 93.
Tracheal intubation should be confirmed by capnography in addition to auscultation. Endobronchial intubation should be promptly recognized and managed. In the event of failure to intubate the trachea after rapid sequence induction, it is imperative to institute a failed intubation drill without delay. Repeated attempts and a second dose of suxamethonium are seldom beneficial and often detrimental. The primary objective in the management of failed intubation is to ensure adequate maternal oxygenation despite the concerns of foetal wellbeing or risk of regurgitation 17.
Patients will need suitable ventilators for adequate ventilation. They need large tidal volumes of 10–12 mls/kg and positive end expiratory pressure (PEEP) may be avoided 25, as though it increases partial pressure of oxygen in blood ( PaO2 ) 25, it might decrease cardiac output and oxygen delivery to foetus 25. Extubation must be done when awake in left lateral position or semi upright position after adequate reversal of muscle relaxant
Antibiotic prophylaxis is a must as high incidence of wound infection in these patients 46, 47. There is increased risk of post operative respiratory failure and hence morbidly obese parturients are best managed in intensive care management or high dependency care post operatively after general anaesthesia 75. Adequate pain control (Patient controlled analgesia / patient controlled epidural analgesia ( PCA/PCEA) to assure post op deep breathing. Infiltrative analgesia at the end of surgery can be carefully used to decrease requirement of post op analgesia. Post operative oxygen should be given and continuous positive airway pressure if required.
Thromboprophylaxis should be given after liasing with the obstetricians as to the dose and frequency required. Both pharmacological and mechanical methods and early mobilization should be used for thromboprophylaxis. It has been suggested that low molecular weight heparin (LMWH) dosing should be based on actual body weight 94.
The anticoagulation status of the patient becomes particularly important for the anesthesiologist when the patient has a spinal or an epidural catheter. According to European guidelines (when a single daily dosing of low molecular weight heparin (LMWH's) is used), catheters can be removed 10–12 hrs after the last dose of low molecular weight heparin (LMWH) and 4 hrs before the next dose.
Subcutaneous and Intramuscular routes of drug administration should be avoided as they are less reliable.
Seidell JC. Epidemiology of obesity. Semin Vasc Med 2005; (1):3–14.
Friedman N et al. Overweight and obesity: an overview of prevalence, clinical impact, and economic impact. Dis Manag 2004; 7 (Suppl 1):S1–S6.
Malecka-Tendera E et al. Childhood obesity: a pandemic of the twentyfirst century. Int J Obes 2006; 30 (Suppl 2):S1–S3.
Deitel M. Some consequences of the global obesity epidemic. Obes Surg 2005; 15:1–2.
National Institutes of Health Consensus Development Conference Statement. Health implications of obesity. Ann Int Med 1985; 103: 147–51
Bray GA ( 1985 ) complications of obesity. Ann Intern Med 103:1052 – 1062
M Tanaka et al – Anesthetic management in obese parturient, J of Anesthesia (1999), 13:217 - 229
Kim SY, Dietz PM, England L et al. Trends in pre-pregnancy obesity in nine states, 1993–2003. Obesity (Silver Spring) 2007; 15: 986–93.
World Health Organization. Obesity: Preventing and Managing the Global Epidemic. Report on a WHO consultation, WHO Technical Report Series 894. Geneva: WHO, 2000
Department of Health, London. Health survey for England 2002 – trends [WWW document].
Naimark A, Cherniak R. Compliance of the respiratory system and its components in health and obesity. J Appl Physiol 1960; 15: 377–82.
Unterborn J. Pulmonary function testing in obesity, pregnancy, and extremes of body habitus. Clin Chest Med 2001; 22: 759–67.
Eng M, Butler J, Bonica JJ. Respiratory function in pregnant obese women. Am J Obstet Gynecol 1975; 123: 241–5.
Cullen JH, Formel PF. The respiratory defects in extreme obesity. Am J Med 1962; 32: 525–31.
Lee JJ, Larsen RH, Buckley JJ et al. Pulmonary function and its correlation to the degree of obesity in 294 patients. Anesth Rev 1981; 8: 28–32.
Vaughan RW, Cork RC, Hollander D. The effect of massive weight loss on arterial oxygenation and pulmonary function tests. Anesthesiology 1981; 54: 325–8.
Saravanakumar K etal. Obesity and obstetric anaesthesia. Anaesthesia 2006; 61:36–48.
Stephansson O et al. Maternal weight, pregnancy weight gain and the risk of antepartum stillbirth. American Journal of Obstetrics and Gynecology 2001; 184: 463–9.
Veille JC, Hanson R. Obesity, pregnancy, and left ventricular functioning during the third trimester. Am J Obstet Gynecol 1994; 171: 980–3.
Vasan RS. Cardiac function and obesity. Heart 2003; 89: 1127–9.
Tomoda S, Tamura T, Sudo Y et al. Effects of obesity on pregnant women: maternal hemodynamic change. Am J Perinat 1996; 13: 73–8.
J Mhyre – anaesthetic management of morbidly obese pts
Tsueda K et al, Obesity supine death syndrome: reports of two morbidly obese patients. Anesthesia and Analgesia 1979; 58: 345–7.
Mendelson CL. The aspiration of stomach contents into the lungs during obstetric anesthesia. Am J Obstet Gynaecol 1945; 52: 191–204.
Olsson GL, Hallen B, Hambraeus-Jonzon K. Aspiration during anaesthesia: a computer-aided study of 185358 anaesthestics. Acta Anaesthesiol Scand 1986; 30: 84–92.
Brock Utne JG, Dow TG et al gastric and lower oesophageal pressure in early pregnancy. Br J Anaestheisa 1981; 53: 381 -384
Obrien TG Jr; LOS pressure and oesophageal function in obese humans J clin gastroenterology. 1980; 2:145 -148
Roberts RB, Shirley MA – reducing the risk of acid aspiration during caesarean section, anaesthe analgesia 1974; 53: 859 - 868
A . Casati , M . Putzu, Anesthesia in the obese patient: Pharmacokinetic considerations . Journal of Clinical Anesthesia , Volume 17 , Issue 2 , Pages 134 - 145
Hodgkinson R, Husain FJ. Obesity and the cephalad spread of analgesia following epidural administration of bupivacaine for cesarean section. Anesth Analg 1980; 59: 89–92.
Hogan QH, Prost R, Kulier A et al. Magnetic resonance imaging of cerebrospinal fluid volume and the influence of body habitus and abdominal pressure. Anesthesiology 1996; 84: 1341–9.
Hackney JD, Crane MG, Collier CC et al. Syndrome of extreme obesity and hypoventilation: studies of etiology. Ann Int Med 1960; 51: 541–52.
Greene NM. Distribution of local anesthetic solutions within the subarachnoid space. Anesth Analg 1985; 64: 715–30.
Greene NM Distribution of local anesthetics within the subarachnoid space. Anesthesia and Analgesia 1985;64:715 -30
Maternal obesity and pregnancy outcome: a study of 287 213 pregnancies in London, N J Sebire et al International Journal of Obesity (2001) 25, 1175 - 82
Kumar AS – Pregnancy outcome in women with morbid obesity J Gynaecology Obstetric 97 (2001) 101
Gross T etall - Obesity in pregnancy : risks and outcome. Obstet Gynaecol 56 ( 1980 ) 446
Johnson SR et al. Maternal obesity and pregnancy. Surg Gynaecol Obstet 164: 431 -7
Ehrenburg HM et al. The influence of obesity and diabetes on the prevalence of macrosomia. American Journal of Obstetrics and Gynecology 2004; 191: 964–8.
Weiss et all – Obesity, obstret complications and caesarean delivery rate American J Obstetr GYnaec 190 ( 2004 ) 1091 -7
Hood DD et al. Anesthetic and obstetric outcome in morbidly obese parturients. Anethesiology 1993; 79:1210–1218.
Usha Kiran TS et al. Outcome of pregnancy in a woman with an increased body mass index. British Journal of Obstetrics and Gynaecology 2005; 112: 768–72.
Myles TD et al. Obesity as an independent risk factor for infectious morbidity in patients who undergo cesarean delivery. Obstetrics and Gynecology 2002; 100: 959– 64.
Andreasen KR et al. Obesity and pregnancy. Acta Obstetricia et Gynecologica Scandinavica 2004; 83: 1022–9.
Galtier-Dereure F et al. Obesity and pregnancy: complications and cost. American Journal of Clinical Nutrition 2000; 71 (Suppl. ): S1242–8.
Obesity in pregnancy. Yu CK, Teoh TG, Robinson S BJOG. 2006 Oct;113(10):1117-25. Epub 2006 Aug 10
Watkins ML et al. Maternal obesity and risk for birth defects. Pediatrics 2003; 111: 1152–8.
Stephansson O et al. Maternal weight, pregnancy weight gain and the risk of antepartum stillbirth. American Journal of Obstetrics and Gynecology 2001; 184: 463–9.
Cedergren Mi : Maternal morbid obesity and risk of adverse pregnancy outcome Obstetr Gynecol 103 (2004) 219
Weiss JL, Malone FD, Emig D et al. Obesity, obstetric complications and cesarean delivery rate – A population-based screening study. Am J Obstet Gynecol 2004; 190: 1091–7.
Munnur U et al , Airway problems in pregnancy. Crit Care Med 2005; 33 (10 Suppl):S259–S268.
Juvin P, et al. Difficult tracheal intubation is more common in obese than lean patients. Anesthesia and Analgesia 2003; 97: 595 – 600.
D’Angelo R, Dewan DD. Obesity. In: Chestnut DH, ed. Obstetric Anesthesia: Principles and Practice, 2004: 893–903
Merah NA et al. Prediction of difficult laryngoscopy in a population of Nigerian obstetric patients. West African Journal of Medicine 2004; 23: 38–4.
Rocke DA et al,. Relative risk analysis of factors associated with difficult intubation in obstetric anesthesia. anesthesiology 1992; 77: 67–73.
Bell RL et al. Postoperative considerations for patients with obesity and sleep apnea. anaesthesiol clin N Am 2005; 23:493–500
Shankar KB et all – airway changes during pregnancy 1997 – anaesthesiology 87: A895
ACOG committee opinion number 315. Obesity in pregnancy – Obstetr Gynaecol 2005; 106:671 -75
Lean ME. Obesity and cardiovascular disease: the waisted years. British Journal of Cardiology 1999; 6: 269–73.
Hess PE, Pratt SD, Lucas TP et al. Predictors of breakthrough pain during labor epidural analgesia. Anesth Analg 2001; 93: 414–8.
Howell CJ. Epidural versus non-epidural analgesia for pain relief in labour (Cochrane Review). In: The Cochrane Library, Issue 4. Chichester: John Wiley & Sons, Ltd., 2004.
Von Ungern-Sternberg BS et al, The effect of epidural analgesia in labour on maternal respiratory function. Anaesthesia 2004; 59: 350–3.
Cascio M, et al. Labour analgesia with intrathecal fentanyl decreases maternal stress. Canadian Journal of Anaesthesia 1997; 44: 605–9.
Patel J. Anaesthesia for LSCS in a morbidly obese patient. Anaesthesia and Intensive Care 1999; 27: 216–9.
Chetty V et al - Difficult spinal anaesthesia for caesarean section in two obese pregnant patients. West African Journal of Medicine 2001; 20: 274–6.
Jordan H, Perlow MD, Mark A, Morgan MD. Massive maternal obesity and perioperative cesarean morbidity. American Journal of Obstetrics and Gynecology 1994; 170: 560–5.
Vincent RD, Chestnut DH. Which position is more comfortable for the parturient during identification of the epidural space? International Journal of Obstetric Anesthesia 1991; 1: 9–11.
Hamza J et al Parturient’s posture during epidural puncture affects the distance from skin to epidural space. Journal of Clinical Anesthesia 1995; 7: 1–4.
Robinson HE, O’Connell CM etall: Maternal outcomes in pregnancies complicated by obesity. Obstet Gynecol 2005; 106: 1357 - 64
Manuel C Vallejo – Anaesthetic management of the morbidly obese parturient; Current opinion in Anaesthesiology 2007, 20: 175 - 180
Clinkscales CP et all An observational study of the relationship between lumbar epidural space depth and BMI in Michigan parturients IJOA 2007;16:323-7
Watts RW. The influence of obesity on the relationship between body mass index and the distance to the epidural space from the skin. Anaesthesia and Intensive Care 1993; 21: 309–10.
Wallace DH, Currie JM. Indirect sonographic guidance for epidural anesthesia in obese pregnant patients. Regional Anesthesia 1992; 17: 233–6.
Grau T, et Ultrasound imaging improves learning curves in obstetric epidural anesthesia: a preliminary study. Canadian Journal of Anesthesia 2003; 50: 1047–50.
Melzack R- Severity of labour pain : influence of physical as w ell psychological variables Can Med Asso J 1984; 130:579 -584
Lewis G (ed). Confidential Enquiries into Maternal and Child Health. Why Mothers Die. The Sixth Report of the United Kingdom Confidential Enquiries into Maternal Deaths in the United Kingdom. London. RCOG Press. 2004. www.cemach.org.uk
Rawal N, Holmstrom B, Crowhurst JA et al. The combined spinal–epidural technique. Anesth Clin North Am 2000; 18: 267–95.
Kuczkowski KM, Benumof JL. Repeat Caesarean section in a morbidly obese parturient: a new anaesthetic option. Acta Anaesthesiologica Scandinavia 2002: 46: 753 – 4
van de Velde M, Teunkens A, Hanssens M et al. Post dural puncture headache following combined spinal epidural or epidural anesthesia in obstetric patients. Anaesth Intensive Care 2001; 29: 595–9.
Pan PH, Bogard TD, Owen MD. Incidence and characteristics of failures in obstetric neuraxial analgesia and anesthesia: a retrospective analysis of 19,259 deliveries. Int J Obstet Anesth 2004; 13: 227–33.
Norris MC. Are combined spinal–epidural catheters reliable? Int J Obstet Anesth 2000; 9: 3–6.
Eappen S, Blinn A, Segal S. Incidence of epidural catheter replacement in parturients: a retrospective chart review. Int J Obstet Anesth 1998;7: 220–5.
Coker LL Continuous spinal anaesthesia for Caesarean section for a morbidly obese patient: a case report. AANA J 2002;70: 189 -192
Faure E et al Incidence of postdural puncture headache in morbidly obese parturients. Reg Anesth 1994; 19: 361-363
Collins JS, Lemmens HJ, Brodsky JB et al. Laryngoscopy and morbid obesity: a comparison of the 'sniff' and 'ramped' positions. Obes Surg 2004; 14: 1171–5.
Baraka AS, Taha SK, Aouad MT et al. Preoxygenation – comparison of maximal breathing and tidal volume breathing techniques. Anesthesiology 1999; 91: 612–6.
Bray GA ( 1985 ) complications of obesity. Ann Intern Med 103:1052 – 1062
Michota F, Merli G. Anticoagulation in special patient populations: are special dosing considerations required? Clev Clin J Med 2005; 72 (Suppl. 1): S37–42.
The aim of this article is to review the diagnosis and management of Dementia with Lewy Bodies. Dementia with Lewy bodies (DLB) is considered the second most common cause of dementia in the elderly after Alzheimer's disease. Diagnostic criteria for DLB is categorised into central feature ( progressive dementia), core features( fluctuating cognition, recurrent visual hallucinations and parkinsonism), suggestive features( rapid eye movement sleep behaviour disorder, increased sensitivity to neuroleptics and low dopamine transporter uptake in the brain's basal ganglia) and supportive features(repeated falls , transient loss of consciousness, hallucinations in other modalities, visuospacial abnormalities and autonomic dysfunction). DLB patients have the diffuse presence of Lewy Bodies in both sub cortical and cortical areas of the brain. Patients with DLB also have more severe dopamine and acetylcholine loss as compared to Alzheimer’s disease. Cholinesterase inhibitors can be used for the treatment of neuropsychiatric symptoms. Treatment with levodopa-carbidopa combinations should be considered when parkinsonian symptoms cause functional impairment. Antipsychotics should be used with great caution due to increased extra pyramidal adverse reactions. Clonazepam can be helpful to manage REM sleep behaviour disorder.
Clinicians need to be aware of the diagnosis of DLB in order to provide appropriate pharmacological and nonpharmacological treatment for its cognitive, neuropsychiatric, motor and sleep disturbances without causing distressing side effects due to inappropriate drug prescription.
Dementia with Lewy bodies (DLB) is considered the second most common cause of dementia in the elderly after Alzheimer's disease. DLB is a progressive neurological disorder characterized by core features of cognitive impairment, psychosis and Parkinsonism. The disease is commonly referred to by a number of names, such as Lewy Body Disease, Lewy Body dementia, dementia with Lewy Bodies, or diffuse Lewy Body Disease. Prevalence estimates of DLB, depending on case criteria, range from0 to 5% with regard to the general population, and from 0 to30.5% of all dementia cases 1. It is claimed that DLB accounts for 20% of late onset dementia 2, 3 .Most studies suggest that DLB is slightly more common in men than in women. DLB is a disease of late middle age and old age. DLB has been described in Asian, African, and European races.
Friederich Lewy discovered abnormal proteins called Lewy Bodies in early1900’s These Lewy Body proteins are spherical intraneuronal cytoplasmic inclusions 15-30um in diameter and are found in the brainstem of patients with Parkinson’s disease. In DLB, these abnormal proteins are found diffusely throughout other areas of the brain including midbrain and the cerebral cortex. The brain chemical acetylcholine is depleted, causing disruption of perception, thinking, and behaviour. Dementia with lewy bodies shares characteristics with both Alzheimer's disease and Parkinson's disease. This can lead to difficulty or delay in reaching the right diagnosis of DLB.
Clinical features
First consensus guidelines for diagnosis of DLB were published in 1996 4 and reviewed in 1999 5.The latest consensus diagnostic criteria for DLB was agreed in the third report of the DLB consortium in 2005 6.
Diagnostic criteria for Dementia with lewy bodies 4-7
Central feature
·Progressive dementia - deficits in attention and executive function are typical. Prominent memory impairment may not be evident in the early stages.
Core features:
·Fluctuating cognition with pronounced variations in attention and alertness.
·Recurrent complex visual hallucinations
·Spontaneous features of Parkinsonism.
Suggestive features:
·REM sleep behaviour disorder (RBD), which can appear years before the onset of dementia and Parkinsonism.
·Severe sensitivity to neuroleptics occurs in up to 50% of LBD patients who take them.
·Low dopamine transporter uptake in the brain's basal ganglia as seen on SPECT and PET imaging scans.
Supportive features:
·Repeated falls and syncope (fainting).
·Transient, unexplained loss of consciousness.
·Autonomic dysfunction.
·Hallucinations of other modalities.
·Visuospatial abnormalities like depth perception, object orientation, directional sense and illusions
·Other psychiatric disturbances like systematized delusions, aggression and depression.
A probable LBD diagnosis requires either:
·Dementia plus two or more core features, or
·Dementia plus one core feature and one or more suggestive features.
A possible LBD diagnosis requires:
·Dementia plus one core feature, or
·Dementia plus one or more suggestive features.
Data from 4-7
COGNITIVE IMPAIRMENT
Prominent memory impairment may not be evident in the early stages.Cognitive features distinguishing DLB from AD are more prominent impairment of attention, executive functioning (e.g., planning, prioritizing, sequencing), and visuospatial problems (such as problems in following an unfamiliar route) 8, 9. Mental inflexibility, perseveration, and intrusion are more likely with DLB than with AD 10. Patients with DLB have more difficulties in clock drawing or figure copying as compared to patients with Alzheimer’s disease who have more prominent memory changes on mini mental state examination 8, 11-13. A core feature of DLB is the fluctuation in cognitive performance, which can occur early in the illness. By way of example, one day a patient may be able to hold a sustained conversation, the next they may be drowsy, inattentive and almost mute.
Visual Hallucinations
Visual Hallucinations are another core feature distinguishing DLB from AD. In DLB, hallucinations are typically recurrent, well formed, and complex and are usually detailed. Patients may see images of people or animals that they recognise. Some patients see coloured patterns or shapes. Presence of hallucinations with substantial fluctuation in attention can lead clinicians to diagnose delirium. Hallucinations are not always distressing to patients and many learn to distinguish between real and unreal images: some people actually come to enjoy them. In many patients visual hallucinations are accompanied by delusions which tend to be persecutory in nature.
Parkinsonism
Spontaneous features of Parkinsonism are another core feature of DLB. Patients usually present with rigidity, bradykinesia, gait changes, masklike faces 14, reduced arm swing and a tendency to falls. Resting tremor is less common in DLB than in PD. Development of dementia within 12 months of extrapyramidal signs suggests DLB, whereas late development of dementia makes PD with dementia more likely 4. Patients who have dementia with Lewy bodies tend to respond less favourably to levodopa with carbidopa as compared to patients who have Parkinson's disease with dementia 11, 15.
Others clinical features
Severe sensitivity to antipsychotics occurs in up to 50% of DLB patients who take them, developing Parkinsonism even if they have not shown such signs before drug administration. The associated Parkinsonism is often prolonged, profound and may even be fatal. REM sleep behaviour disorder occurs in about one half of these patients. REM sleep behaviour disorder usually presents with vivid dreams associated with simple or complex motor behaviour during REM sleep 11. Diagnosis of DLB is also supported by repeated falls and syncope, transient loss of consciousness hallucinations in other modalities, visuospacial abnormalities and autonomic dysfunction.
Pathogenesis
The pathology of DLB closely resembles that of Parkinsonism disease. Patients with DLB are characterised by the diffuse presence of Lewy Bodies in both subcortical and cortical areas of the brain whereas Parkinson’s disease patients have lewy bodies in the subcortical areas of the brain mainly substantia nigra and locus cerules 11, 16. Both DLB and Parkinson’s disease are associated with abnormal aggregation of alpha-synuclein which is a nerve terminal protein that is a better marker of lewy bodies than ubiquitin. Biochemically, numerous neurotransmitters, including acetylcholine and dopamine are diminished in DLB. The decrease in acetylcholine may be more severe than in Alzheimer’s disease.
Pathological features in DLB 17, 18
Diffuse Lewy bodies - Essential for diagnosis of DLB Lewy neuritis Senile Plaques (all morphological types) Neurofibrillary tangles Neuronal loss in substantia nigra Neuronal loss in locus coeruleus Meynert nucleus neuronal loss Microvacuolation and synapse loss Neurochemical abnormalities and neurotransmitter deficits e.g. Ach, Dopamine Data from 17, 18
Differential Diagnosis
DLB can be easily confused with Alzheimer’s disease (AD) and Parkinson’s disease (PD).It is important to differentiate between DLB, AD and PD due to differences in treatment approaches. As compared to AD, patients suffering from DLB more frequently show signs of frontal lobe dysfunction, more prominent visual and auditory hallucinations, fluctuating cognitive performance, greater sensitivity to neuroleptics 19 and parkinsonian symptoms. Patients with DLB also have more severe dopamine and acetylcholine loss as compared to AD. DaT FP-CIT scan can be useful to differentiate between DLB and AD. Other diagnoses which can be confused with DLB include delirium and psychiatric illnesses.
Differential Diagnosis of DLB 20
Alzheimer’s Diseas Parkinson’s Disease Dementia in Parkinson’s Disease Psychiatric illnesses like mania and psychotic depression Vascular Dementia Delirium
Investigations
It is important to do dementia screen to rule out any reversible causes of cognitive impairment.
Blood tests
Laboratory studies should include those usually ordered in a dementia evaluation 21, including the following:
·FBC, ESR, CRP, biochemical screen
·Urea and creatinine
·T4 and TSH
·Glucose
·B12 and folate
·Clotting & albumin
·Syphilis serology
·HIV - if in young person
·Caeruloplasmin
Urine tests
Perform a midstream urine test if delirium is a possibility.
Imaging studies
·Structural imaging can be used to exclude other cerebral pathologies and help establish the subtype of dementia. Imaging studies may help to identify treatable causes such as subdural haematoma, normal pressure hydrocephalus, and cerebral tumours.
·Brain MRI is indicated to distinguish DLB from vascular dementia. Patients with vascular dementia often have white matter lesions on MRIs, whereas patients with DLB do not.
·Regionally distinct patterns of hypoperfusion on single-photon emission computed tomography (SPECT) or hypometabolism on positron emission tomography (PET) can help differentiate Frontotemporal Dementia, AD and Vascular Dementia, and dopaminergic loss in the basal ganglia can differentiate DLB from AD 22.
·Reduced dopamine transporter activity in the basal ganglia is seen with positron emission tomography (PET) scanning or single-photon emission CT (SPECT) scanning.
·DaTSCAN (Ioflupane, 123-I FP-CIT) SPECT imaging.DaTSCAN contains Ioflupane labelled with radioactive iodide in an ethanolic solution. DaTSCAN is a drug used as part of a diagnostic procedure called SPECT imaging. DaTSCAN SPECT is indicated for detecting loss of functional dopaminergic neuron terminals in the striatum. The sensitivity of the FP-CIT scan for the diagnosis of DLB is 88% and specificity is 100 % 23.It helps to differentiate probable dementia with Lewy bodies from Alzheimer’s disease.
Management
There is limited evidence about specific interventions but available data suggests a role for cholinesterase inhibitors, atypical antipsychotics, levodopa and clonazepam. For the treatment of agitation and hallucinations associated with DLB, acetyl cholinesterase inhibitors are the drugs of choice. In a small minority of patients, motor features are worsened with cholinesterase inhibitors. Most experts recommend atypical neuroleptics when cholinesterase inhibitors are ineffective. Levodopa/carbidopa may improve motor function in some patients with DLB; however, in many patients this combination has no effect and may exacerbate psychiatric symptoms or confusion. Depression is frequent in DLB patients and may result from damage in the dorsal raphe and locus ceruleus and/or as a psychological response to impaired function. Selective serotonin reuptake inhibitors are the drugs of choice.
Pharmacological Treatment
Acetyl cholinesterase inhibitors
Cholinergic deficits in DLB are even more severe than in AD 24. Patients with DLB are more likely to improve with cholinesterase inhibitor therapy. Encouraging results have been obtained with Rivastigmine, Donezepil and galantamine. Double-blinded, placebo-controlled studies 25-27 have demonstrated that rivastigmine may decrease neuropsychiatric symptoms associated with DLB, particularly apathy, anxiety, hallucinations, and delusions. There is also some evidence from several case reports, open label trials and case series about the use of acetyl cholinesterase inhibiters including Rivastigmine and Donepezil in DLB 28-32.
Atypical neuroleptics
Due to increased sensitivity to antipsychotics, clinicians are generally cautious about the use of these drugs in patients with DLB. There have been multiple studies about the use of atypical antipsychotics like risperidone, olanzapine and quetiapine in DLB patients for the management of neuropsychiatric symptoms 33-37. Patients with DLB frequently have distressing neuropsychiatric symptoms. When these symptoms are mild, no medical treatment may be necessary. Acetyl cholinesterase inhibitors should usually be tried first to treat neuropsychiatric symptoms 38. Atypical antipsychotics appear to be better tolerated by DLB patients 39. Most experts recommend atypical neuroleptics when cholinesterase inhibitors are ineffective. Neuroleptics should be reserved for situations where the psychosis is causing serious distress or putting the patient or others at risk. Very slow titration of the neuroleptic medication is indicated.
Anti-Parkinson’s Medications Patients with DLB can have troublesome parkinsonian symptoms which might need treatment. Treatment with levodopa-carbidopa combinations should be considered when symptoms cause functional impairment. Most of the evidence for benefit comes from case series40, 41.
Benzodiazepines Clonazepam can be helpful in treating REM sleep behaviour disturbances in DLB patients 42, 43. Antidepressants Patients with DLB have increased frequency of depression and anxiety. Selective serotonin reuptake inhibitors (SSRI’s) are the drugs of choice.
NonPharmacological Treatment
Nonpharmacological management mainly involves education of the patient and carers to deal with specific symptoms of the illness as well as general issues of caring for a patient with dementia 20.Various interventions including education of patient and family, structuring of environment, teaching behavioral skills and improving sensory impairment have been found useful in other types of dementias and might also be useful in patients suffering from dementia with lewy bodies 44-48.
COMPETEING INTERESTS:
None declared
AUTHOR DETAILS
JAVED LATOO, DPM, MRCPsych. Specialty Registrar (ST5) in Psychiatry with special interest in Neuropsychiatry, Royal Free and University College London, London, United Kingdom FARIDA JAN, MRCPsych. Specialty Registrar in Old Age Psychiatry, Eastern Deanery, United Kingdom
CORRESSPONDENCE: Dr Javed Latoo, North East London NHS Foundation Trust, Mascalls Park Hospital, Mascalls Lane Brentwood, Essex, CM145HQ, United Kingdom
Email: javedlatoo@googlemail.com
REFERENCES
1.Julia Zaccai, Cherie McCracken and Carol Brayne. A systematic review of prevalence and incidence studies of dementia with Lewy bodies Age and Ageing 2005 34(6):561-566.
2.Campbell S, Stephens S, Ballard C, Dementia with Lewy bodies: Clinical features and treatment, Drugs Aging, 2001; 18:397–407.
3.McKeith IG, Spectrum of Parkinson’s disease, Parkinson’s dementia, and Lewy body dementia, Neurol Clin, 2000; 18: 865–902.
4.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. Nov 1996; 47(5):1113-24.
5.McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. Sep 22 1999; 53(5):902-5.
6.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. Dec 27 2005; 65(12):1863-72.
7.Lewy Body Dementia Association, Inc. Information page accessed online on 23 Aug. 08 at http://www.lbda.org/category/3438/symptoms.htm
8.Ballard C, Ayre G, O’Brien J, Sahgal A, McKeith IG, Ince PG, et al. Simple standardised neuropsychological assessments aid in the differential diagnosis of dementia with Lewy bodies from Alzheimer’s disease and vascular dementia. Dement Geriatr Cogn Disord 1999; 10:104-8.
9.Mori E, Shimomura T, Fujimori M, Hirono N, Imamura T, Hashimoto M, et al. Visuoperceptual impairment in dementia with Lewy bodies. Arch Neurol 2000; 57(4):489-93.
10.Doubleday EK, Snowden JS, Varma AR, Neary D. Qualitative performance characteristics differentiate dementia with Lewy bodies and Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2002; 72:602-7.
11.McKeith I, Mintzer J, Aarsland D, Burn D, Chiu H, Cohen-Mansfield J, et al. Dementia with Lewy bodies. Lancet Neurol 2004; 3:19-28.
12.Knopman DS, Boeve BF, Petersen RC. Essentials of the proper diagnoses of mild cognitive impairment, dementia, and major subtypes of dementia. Mayo Clin Proc 2003; 78:1290-308.
13.Mosimann UP, McKeith IG. Dementia with Lewy bodies-diagnosis and treatment. Swiss Med Wkly 2003; 133:131-42.
14.Aarsland D, Ballard C, McKeith IG, Perry RH, Larsen JP. Comparison of extrapyramidal signs in dementia with Lewy bodies and Parkinson’s disease. J Neuropsychiatry Clin Neurosci 2001; 13(3):374-9.
15.Stewart JT. Defining diffuse Lewy body disease. Tetrad of symptoms distinguishes illness from other dementias. Postgrad Med 2003; 113:71-5.
16.NussbaumRL, Ellis CE.Alzheimer’s disease and Parkinson’s disease {Published correction appears in N Eng J Med 2003; 348:2588}.N Engl J Med 2003; 348: 1356-64.
17.Alex Mitchell, Textbook of Neuropsychiatry and Behavioural Neurology Saunders 2004.
18.Paul G. Ince, Elaine K. Perry, Chris. M. Morris. Dementia with Lewy Bodies. A Distinct Non-Alzheimer Dementia Syndrome? Brain Pathology. April 1998.
19.Bolla LR, Filley CM, Palmer RM, Dementia DDx: Office diagnosis of the four major types of dementia, Geriatrics, 2000; 55:34–46.
20.Frank Christopher, Dementia with Lewy bodies, Can Fam Physician 2003; 49:1304-1311.
21.NICE (November 2006). Dementia guidelines
22.O'Brien JT. Role of imaging techniques in the diagnosis of dementia.Br J Radiol. 2007 Dec;80 Spec No 2:S71-7
23.Walker Z et al. Dementia with Lewy bodies: a comparison of clinical diagnosis, FP-CIT SPECT imaging and autopsy.J Neurology Neurosurgery Psychiatry.2007 Nov;78(11):1176-81
24.Gomez-Tortosa E, Ingraham AO, Irizarry MC, Hyman BT. Dementia with Lewy bodies. J Am Geriatr Soc 1998; 46:1449-58.
25.Wesnes K, McKeith IG, Ferrara R, Emre M, Del Ser T, Spano PF, et al. Effects of rivastigmine on cognitive function in dementia with Lewy bodies: a randomized placebo-controlled international study using the cognitive drug research computerised assessment system. Dement Geriatr Cogn Disord 2002; 13:183-92.
26.McKeith IG, Grace J, Walker Z, Byrne EJ, Wilkinson D, Stevens T, et al. Rivastigmine in the treatment of dementia with Lewy bodies: preliminary findings from an open trial. Int J Geriatr Psychiatry 2000; 15:387-92.
27.McKeith IG, Del Ser T, Spano PF, Emre M, Wesnes K, Anand R, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000; 356(9247):2031-6.
28.Maclean LE, Collins CC, Byrne EJ. Dementia with Lewy bodies treated with rivastigmine: effects on cognition, neuropsychiatric symptoms, and sleep. Int Psychogeriatr 2001; 13(3):277-88.
29.Grace J, Daniel S, Stevens T, Shankar KK, Walker Z, Byrne EJ, et al. Long-term use of rivastigmine in patients with dementia with Lewy bodies: an open-label trial. Int Psychogeriatr 2001; 13(2):199-205.
30.Samuel W, Caligiuri M, Galasko DR, Lacro J, Marini M, McClure FS, et al. Better cognitive and psychopathologic response to donepezil in patients prospectively diagnosed as dementia with Lewy bodies: a preliminary study. Int J Geriatr Psychiatry 2000; 15:794-802.
31.Querfurth HW, Allam GJ, Geffroy MA, Schiff HB, Kaplan RF. Acetylcholinesterase inhibition in dementia with Lewy bodies: results of a prospective pilot trial. Dement Geriatr Cogn Disord 2000;11:314-21.
32.Shea C, MacKnight C, Rockwood K. Donepezil for treatment of dementia with Lewy bodies: a case series of nine patients. Int Psychogeriatr 1998; 10(3):229-38.
33.Sechi G, Agnetti V, Masuri R, Deiana GA, Pugliatti M, Paulus KS, et al. Risperidone, neuroleptic malignant syndrome and probable dementia with Lewy bodies. Prog Neuropsychopharmacol Biol Psychiatry 2000; 24(6):1043-51.
34.Kato K, Wada T, Kawakatsu S, Otani K. Improvement of both psychotic symptoms and Parkinsonism in a case of dementia with Lewy bodies by the combination therapy of risperidone and L-DOPA. Prog Neuropsychopharmacol Biol Psychiatry 2002; 26(1):201-3.
35.Fernandez HH, Trieschmann ME, Burke MA, Friedman JH. Quetiapine for psychosis in Parkinson’s disease versus dementia with Lewy bodies. J Clin Psychiatry 2002; 63(6):513-5.
36.Walker Z, Grace J, Oveshot R, Satarasinghe S, Swann A, Katona CL, et al. Olanzapine in dementia with Lewy bodies: a clinical study. Int J Geriatr Psychiatry 1999;14:459-66.
37.Cummings JL, Street J, Masterman D, Clark WS. Efficacy of olanzapine in the treatment of psychosis in dementia with Lewy bodies. Dement Geriatr Cogn Disord 2002; 13:67-73.
39.Andreasen NC, Black DW, Delirium, dementia, and amnestic disorder. In: Introductory Textbook of Psychiatry, 4th ed., Arlington, Virginia: American Psychiatric Publishing, Inc., 2006.
40.Byrne EJ, Lennox G, Lowe J. Diffuse Lewy body disease, clinical features in 15 cases. J Neurol Neurosurg Psychiatry 1989; 47:709-17.
41.Geroldi C, Frisoni GB, Bianchetti A, Trabucci M. Drug treatment in Lewy body dementia. Dement Geriatr Cogn Disord 1997; 8:188-97.
42.Stewart JT. Defining diffuse Lewy body disease. Tetrad of symptoms distinguishes illness from other dementias. Postgrad Med 2003; 113:71-5.
43.Boeve BF, Silber MH, Ferman TJ. REM sleep behavior disorder in Parkinson's disease and dementia with Lewy bodies. J Geriatr Psychiatry Neurol 2004; 17:146-57.
44.Mosimann UP, McKeith IG. Dementia with Lewy bodies-diagnosis and treatment. Swiss Med Wkly 2003; 133:131-42.
45.Burn DJ, McKeith IG. Current treatment of dementia with Lewy bodies and dementia associated with Parkinson's disease. Mov Disord 2003; 18(suppl 6):S72-9.
46.Kalra S, Bergeron C, Lang AE. Lewy body disease and dementia. A review. Arch Intern Med 1996; 156:487-93.
47.Doody RS, Stevens JC, Beck C, Dubinsky RM, Kaye JA, Gwyther L, et al. Practice parameter: management of dementia (an evidence-based review). Report of Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001; 56:1154-66.
48.Neef and Walling. Dementia with Lewy Bodies; An Emerging Disease.Am Fam Physician 2006; 73:1223-9.
Population demographics are changing worldwide, with life expectancy and the proportions of older persons increasing. Older people are the greatest consumers of medications and healthcare resources in developed countries. It is assumed that as more drugs become available and life expectancy continues to increase, the consumption of prescription drugs by older people will increase further and the incidence of potentially inappropriate prescribing will grow. A survey of non-institutionalized older adults in the United States showed an increased usage of all medications with advancing age, the highest prevalence of drug use being in women 65 years of age and older with 12% taking 10 or more medications and 23% taking at least five prescribed drug therapies 1. In most industrialized nations older people consume three times as many prescription medications as younger people and purchase 70% of non-prescription medications 2. In the United States, 12•5% of the population is over 65 years of age but consume 32% of all prescription medications and account for 25% of drug expenditure and 30% of total national healthcare expenditure 3-5. In Ireland, 11•13% of population is over the age of 65 years but consume 47% of all prescription medications 6. In Europe, people over 65 years of age consume on average 2•3 times the amount of health care than do those <65 years of age 7.
POLYPHARMACY:
Polypharmacy has been defined in many different ways and the appropriate definition may differ according to patient population and study setting 9. Fulton and Allen 10 define polypharmacy as: 'the use of medications that are not clinically indicated'. In practice, polypharmacy is defined as using more than a certain number of drugs, irrespective of the appropriateness of drug use 8, 11, 12. Inappropriate prescribing includes the use of medicines that introduce a significant risk of an adverse drug-related event where there is evidence for an equally or more effective but lower-risk alternative therapy available for treating the same condition. Inappropriate prescribing also includes the use of medicines at a higher frequency and for longer than clinically indicated, use of multiple medicines that have recognized drug–drug interactions and drug–disease interactions, and importantly, the under-use of beneficial medicines that are clinically indicated but not prescribed for some reasons. As older patients seek treatment for various ailments from a variety of physicians, they are at increasing risk of accumulating layers of drug therapy. Individuals aged 65 and older use a disproportionate number of prescriptions and over-the-counter medications; 31% use more than one pharmacy and 50% receive prescriptions from more than one prescriber 13. A higher number of primary care physicians and multiple dispensing pharmacies increase the risk of drug–drug interactions 14. The number of medications prescribed to elderly patients, and the complexity of their drug regimens increase over time 15. The potential for an increased risk of drug–drug interactions and adverse drug reactions, and factors such as age-related changes in pharmacodynamics (PD) and pharmacokinetics (PK) must be considered. Diabetes and chronic lung disease predict a greater complexity and cost of drugs regimen in elderly patients with heart failure 16. Besides the increase in diseases and worsening of diseases, the literature also mentions other factors as being responsible for the increase in polypharmacy, i.e. ageing, moving to a residential or nursing home and hospitalization 17, 18. The patient's expectations, the General Practitioner's attitude and consultations with several doctors have been associated with an increase in multiple drug use 19, 20.
EFFECTS OF AGING ON DRUG METABOLISM:
Drug absorption, distribution, metabolism and elimination change as a natural consequence of the ageing process. Changes in drug absorption in older patients may result from decreases in splanchnic blood flow and gastric motility, and increases in gastric pH, and other physiological changes that are associated with ageing. Blood flow and gastric motility may be further diminished by cardiovascular and gastrointestinal drugs used to treat co-morbid conditions. Ageing influences drug excretion. Age-related decreases in glomerular filtration rate are well known. These physiological declines coupled with co-morbid conditions and the use of multiple drugs means that medications eliminated by the renal route requires dose adjustment. Drugs that influence renal function and thus elimination/excretion have the potential to pose serious clinical problems if used concomitantly. With ageing, there is a decrease in lean body mass and total body water with a relative increase in total body fat 21. These changes lead to a decreased volume of distribution for hydrophilic drugs such as lithium, and digoxin where unadjusted dosing can result in higher plasma concentrations, thus increasing the potential for adverse effects. Conversely, lipid soluble drugs such as long-acting benzodiazepines have an increased volume of distribution, thereby delaying their maximal effects and resulting in accumulation with continued use. There is a reduction in hepatic mass and blood flow with ageing 22.
Drugs such as beta-blockers, nitrates and tricyclic anti-depressants that have a first pass effect in the liver may have a higher bioavailability in older people and thus be effective at lower doses. Cytochrome P450 oxidation declines with ageing 24 and drug–drug interactions involving these enzymes are important to recognize. Larger drug storage reservoirs and decreased clearance prolong drug half-lives and lead to increased plasma drug concentrations in older people. If serum albumin is decreased there will be an increase in the active unbound drug concentration for highly protein-bound drugs such as phenytoin, theophylline, warfarin and digoxin. Ageing is also associated with changes in the end-organ responsiveness to drugs at receptor or post-receptor level 25. There is decreased sensitivity to beta-receptors along with a possible decreased clinical response to beta-blockers and beta-agonists 26. Increased sensitivity to drugs such as opiates and warfarin is common 27, 28.
ADVERSE DRUG REACTIONS (ADRs):
The number of elderly is increasing dramatically. In United States, in the next 25 years, as the baby boomer generation begins to turn 65 years old, the number of elderly is expected to double to approximately 70 million. Those older than 85, is now the fastest growing segment of our population. Thus, we can expect the number of adverse drug reactions to increase proportionately. Polypathology, the age-related increase of concurrent diseases, is likely to be the main determinant of drug consumption. However, both over-prescribing and improper prescribing has been reported and seems to contribute to the age-related increase in the prevalence of adverse drug reactions (ADRs) 29, 30. A hospital-based study from Norway showed that the risk of experiencing a drug-related problem increased linearly with the number of drugs on admission 31. A study carried out in the USA found that nursing home patients receiving nine or more drugs were more than twice as likely as patients receiving a lower number of drugs, of experiencing an adverse effect 32. On average, ADRs account for 3%–13% of all the admissions 33-35 and complicate 5%–20% of the stays of patients over 65 years 36-38. More than 40% of persons aged 65 and older use five or more different medications per week, and 12% use 10 or more different medications 39. If an elderly patient takes five or more drugs, he or she has a 35% chance of experiencing an adverse drug event 40. Drug interactions are significant contributors to morbidity 35. Office visits for an adverse drug event increase from 9% of the population per year at age 25–44 years to as high as 56•8% between age 65 and 74 years 41. Inappropriate drug use is one of the risk factors for adverse drug reactions in the elderly. The risk for an adverse drug event is 13% with the use of two medications, but the risk increases to 58% for five medications 42. If seven or more medications are used, the incidence of adverse drug events increases to 82% 42.
INTERVENTIONS:
Older people are a heterogenous group, often with multiple concomitant illnesses and multiple prescriptions. There is a thin line between a healthy old person and an ill old person. Prescribing for older people is challenging as any new medication must be considered in the context of altered pharmacokinetics, altered pharmacodynamics and age-related changes in body composition and physiology. Both over prescription and undue prescription seem to characterize the overall pharmacological therapy of the elderly. Polypharmacy is the main risk factors for ADRs 43. Thus, attempts should be made to curtail inappropriate drug prescription by utilizing different available tools 44. An interdisciplinary medication review of older individuals in the community helps to reduce the cost and number of medications. Polypathology seems the most obvious explanation of the high number of drugs taken by older people, but additional factors deserve consideration. Changes in patient’s medical status over time can cause medications that have been used chronically to become unsafe or ineffective. Particular care must be taken in determining drug dosages and treatment options when prescribing for older adults. “Pill for an ill” approach should be discouraged as many a time pharmacological treatment may carry more adverse effects then the illness itself. Use of electronic medical records and other hand held devices to prescribe appropriate medication doses and check drug to drug interactions has been found useful in reducing the medication related errors and hence adopted by various medical groups and hospital practices. Reviewing medications at every visit is a simple and very helpful tool too especially if patients are encouraged to bring with them a printed list of their current medications (including over the counter drugs). Printing an updated list of the medication changes in bold and large font after a visit with their physician helps patients to follow the recommendations especially in case of geriatric patients who may not remember all the new changes made at an office visit.
CONCLUSIONS:
Polypharmacy is an important issue in the elderly. The problem involves many issues, a number of which have been explored in this article. One of the most important issues involves adverse drug reactions. All pharmaceutical agents have the potential for side effects; therefore, it is obvious that the more drugs one takes the more side effects one will experience. The aging process results in altered metabolism and excretion of medications, and deficits in cognition and senses. Incidence of adverse drug reaction and interactions is increased with polypharmacy. Since adverse drug reactions are a significant cause of morbidity and mortality, as well as an important cause for hospital admissions, minimizing polypharmacy is an important consideration. The general principle of “Start Low and Go Slow” holds true in most scenarios but should be modified to “Start Low, Go Slow but Use Enough” to achieve desired therapeutic effect.




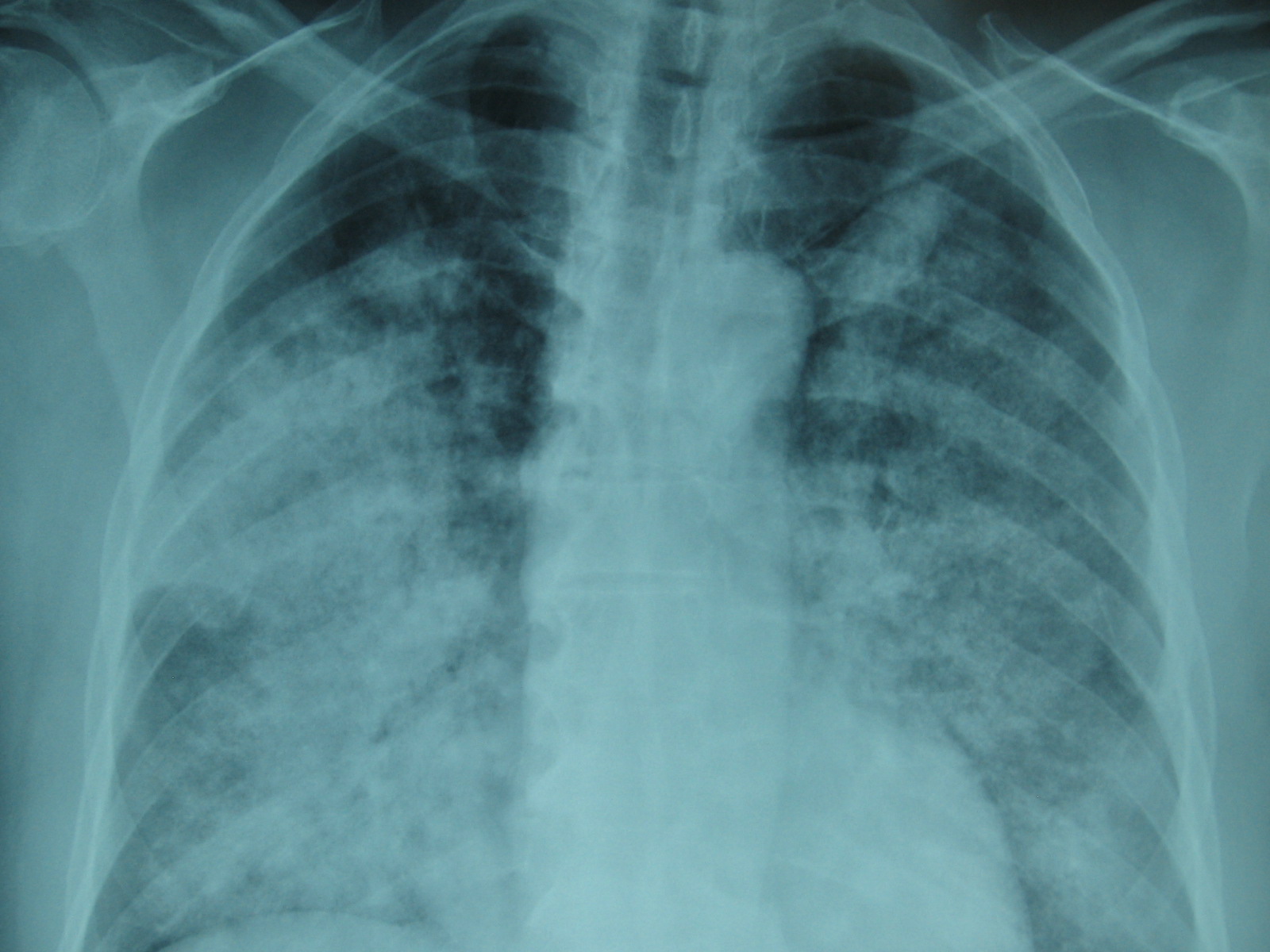
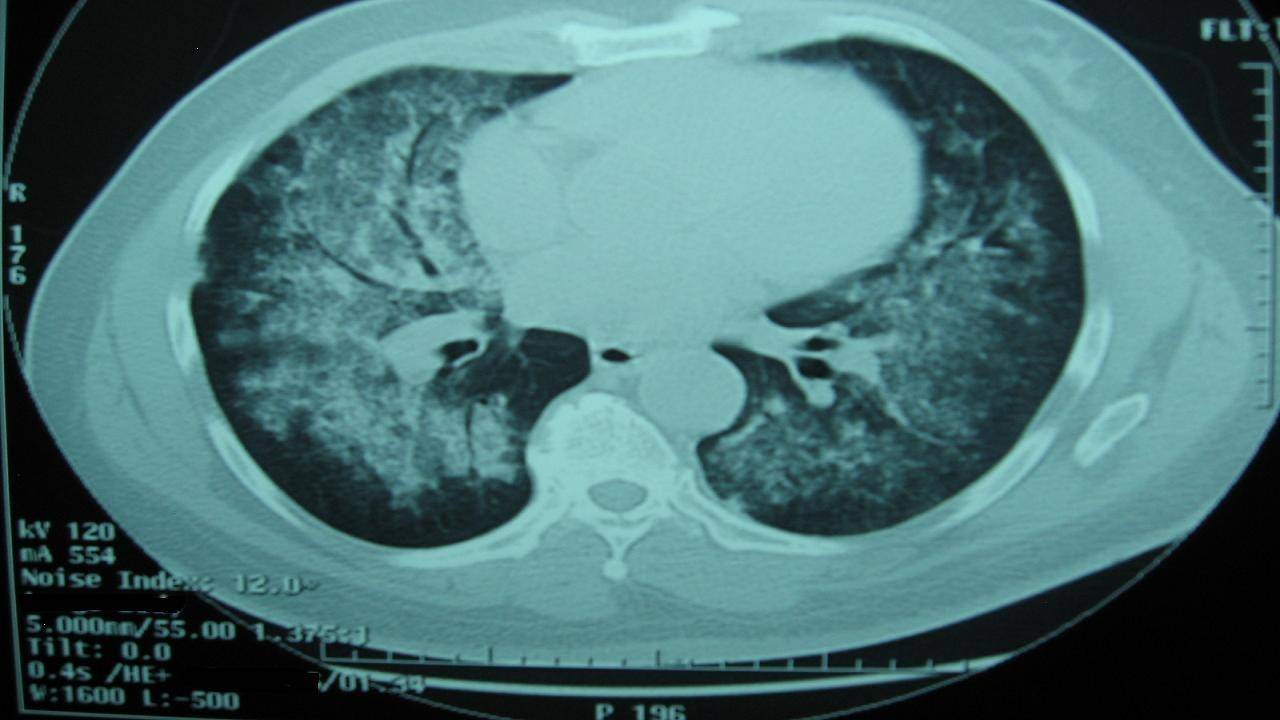

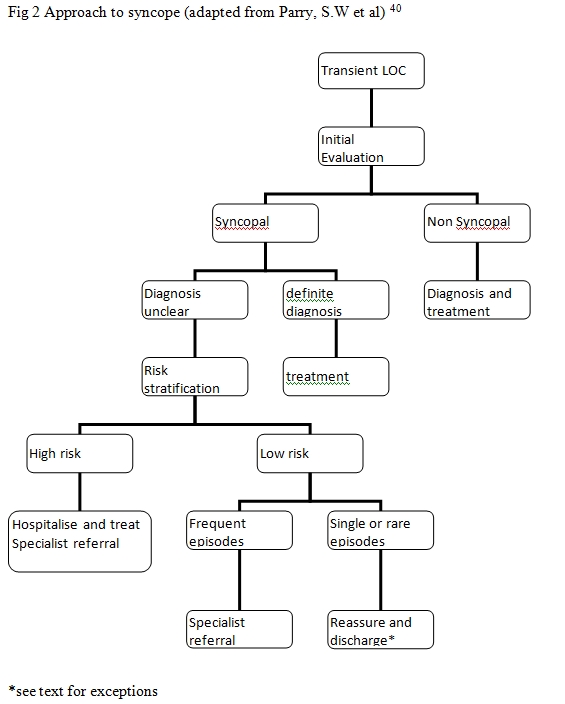 The causes of syncope can be broadly divided in to cardiac causes and non-cardiac causes (Table 1). Initial evaluation leads to a diagnosis in less than 50% patients in most instances4,12-14. If there is uncertainty about diagnosis then the patient is risk stratified. High-risk patients are hospitalised, evaluated and treated whereas early discharge could be considered in low risk patients. Aetiology of Syncope41
The causes of syncope can be broadly divided in to cardiac causes and non-cardiac causes (Table 1). Initial evaluation leads to a diagnosis in less than 50% patients in most instances4,12-14. If there is uncertainty about diagnosis then the patient is risk stratified. High-risk patients are hospitalised, evaluated and treated whereas early discharge could be considered in low risk patients. Aetiology of Syncope41

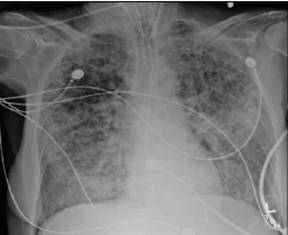
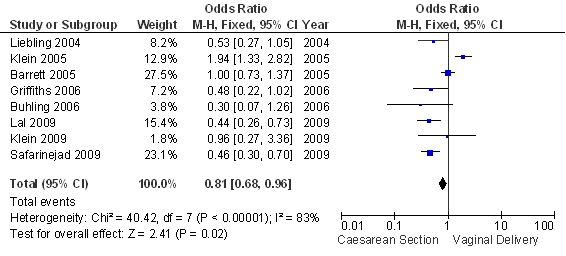
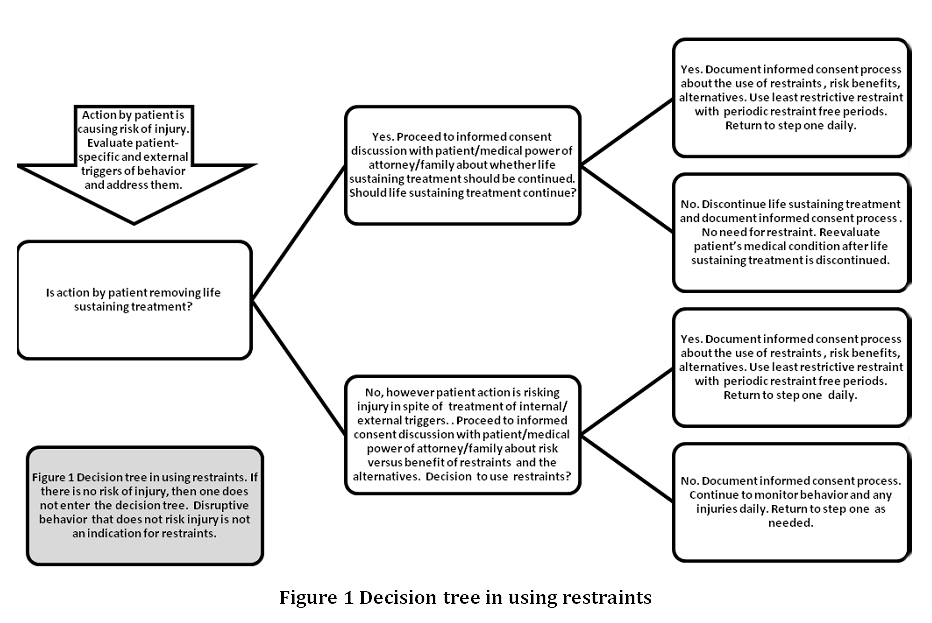
 Figure 1, illustrates a distribution of the peak work-load period which occurred between 9pm-11pm and 1am-3am. This finding contradicts previous surveys done prior to the implementation of the hospital at night where it was noted that the nights were quieter and sub-specialist such as orthopaedic surgeons were not working as hard as general medics (1).
Figure 1, illustrates a distribution of the peak work-load period which occurred between 9pm-11pm and 1am-3am. This finding contradicts previous surveys done prior to the implementation of the hospital at night where it was noted that the nights were quieter and sub-specialist such as orthopaedic surgeons were not working as hard as general medics (1). 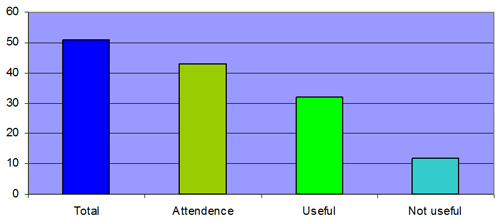 Figure 2, shows the overall experience of the hand-over as judged by the orthopaedic juniors. There was an eighty-five percent (43/51) attendance by the on-call orthopaedic juniors of which seventy-two percent (32/44) thought that it was a useful experience
Figure 2, shows the overall experience of the hand-over as judged by the orthopaedic juniors. There was an eighty-five percent (43/51) attendance by the on-call orthopaedic juniors of which seventy-two percent (32/44) thought that it was a useful experience 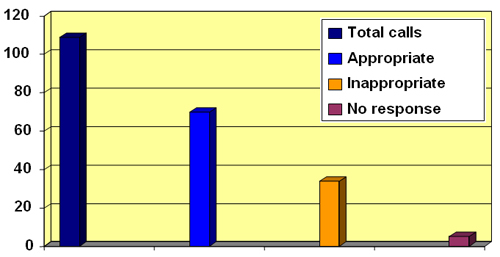 Figure 3 illustrates a distribution of the calls received. As illustrated, 37/109 calls were viewed as inappropriate The majority of the orthopaedic juniors surveyed framed hospital-at-night hand-over was useful but thirty-four percent (37/109) of the calls they received were viewed as inappropriate (Fig 2,3). The most frequent of the inappropriate ward calls were ‘requests to rewrite drug-charts’ (Fig 4,5).
Figure 3 illustrates a distribution of the calls received. As illustrated, 37/109 calls were viewed as inappropriate The majority of the orthopaedic juniors surveyed framed hospital-at-night hand-over was useful but thirty-four percent (37/109) of the calls they received were viewed as inappropriate (Fig 2,3). The most frequent of the inappropriate ward calls were ‘requests to rewrite drug-charts’ (Fig 4,5). 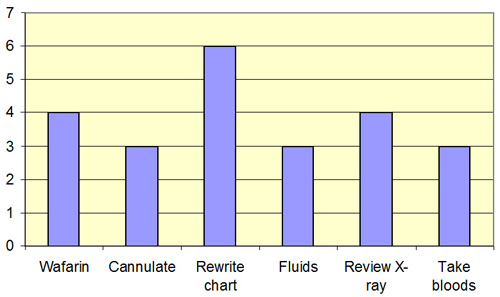 Figure 4 describes the frequencies of these calls. As illustrated the most frequently inappropriate ward-call was to ‘rewrite drug-charts’. There were twenty-three (23/37) inappropriate ward-calls
Figure 4 describes the frequencies of these calls. As illustrated the most frequently inappropriate ward-call was to ‘rewrite drug-charts’. There were twenty-three (23/37) inappropriate ward-calls 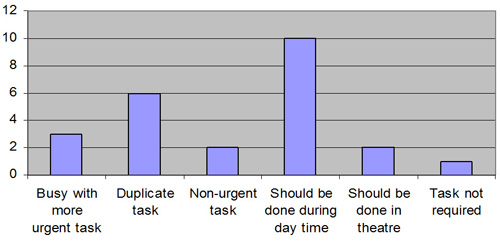 Figure 5 shows the reasons given by the on-call doctors for the inappropriate tasks. The most frequently given reason was that ‘the request should have been done during the day’. The second most frequently given reason for inappropriate calls was the fact that ‘it was a duplicated request’ Our survey also highlighted that the most frequently given reason for inappropriate tasks was firstly that the request should have been done during the day and secondly that it was a duplicated request. These findings seem to defeat the key purpose of the hospital-at-night project for an orthopaedic SHO on-call. Conclusion: Hospital at night cannot function in isolation. There are fewer doctors available then normally and the system has to use their time effectively. 30% of inappropriate calls to wards were to rewrite drug charts and to prescribe warfarin. This should have been identified and performed by the day staff. 10% of calls were to review X-rays. Much of the inappropriate activity could be pre-empted if both day and night staff attended the handover and the tasks outstanding from the day identified. The doctor on the night could then perform an hourly ward round completing these tasks without interrupting at the beginning of the night.
Figure 5 shows the reasons given by the on-call doctors for the inappropriate tasks. The most frequently given reason was that ‘the request should have been done during the day’. The second most frequently given reason for inappropriate calls was the fact that ‘it was a duplicated request’ Our survey also highlighted that the most frequently given reason for inappropriate tasks was firstly that the request should have been done during the day and secondly that it was a duplicated request. These findings seem to defeat the key purpose of the hospital-at-night project for an orthopaedic SHO on-call. Conclusion: Hospital at night cannot function in isolation. There are fewer doctors available then normally and the system has to use their time effectively. 30% of inappropriate calls to wards were to rewrite drug charts and to prescribe warfarin. This should have been identified and performed by the day staff. 10% of calls were to review X-rays. Much of the inappropriate activity could be pre-empted if both day and night staff attended the handover and the tasks outstanding from the day identified. The doctor on the night could then perform an hourly ward round completing these tasks without interrupting at the beginning of the night.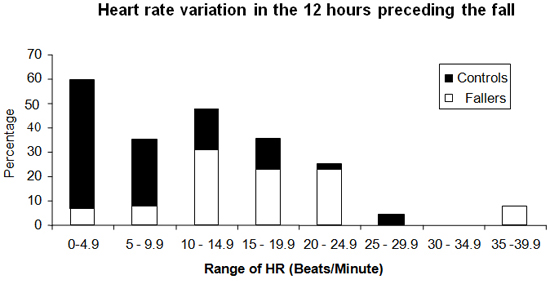
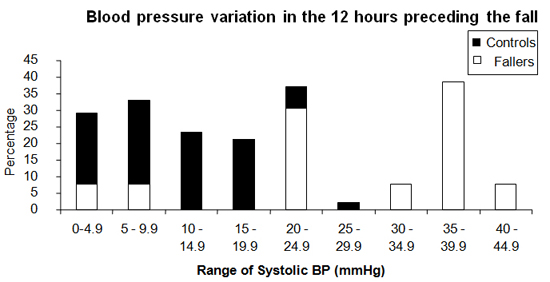
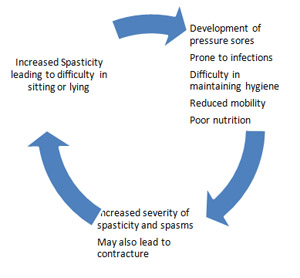 Assessment of spasticity Before any intervention is undertaken to modulate hypertonicity, it is important to attempt to assess the severity of spasticity. Many grading scales are used to quantify spasticity. These address the degree of muscle tone, the frequency of spontaneous spasms and the extent of hyperreflexia. Goniometry, Ashworth scale, Tardieu Scales, Goal attainment scale are only a few of these scales. One of the most widely used scales is the modified Ashworth scale2. Table 2 Modified Ashworth scale
Assessment of spasticity Before any intervention is undertaken to modulate hypertonicity, it is important to attempt to assess the severity of spasticity. Many grading scales are used to quantify spasticity. These address the degree of muscle tone, the frequency of spontaneous spasms and the extent of hyperreflexia. Goniometry, Ashworth scale, Tardieu Scales, Goal attainment scale are only a few of these scales. One of the most widely used scales is the modified Ashworth scale2. Table 2 Modified Ashworth scale